




El tratamiento de la aspergilosis invasora exige la utilización de algunos fármacos que de forma característica presentan propiedades farmacocinéticas complejas, cuyo conocimiento es imprescindible para alcanzar la máxima eficacia con el mínimo riesgo para el paciente. Las formulaciones lipídicas de anfotericina B son muy distintas en su comportamiento farmacocinético, con concentraciones plasmáticas de la forma liposómica muy elevadas en probable relación con la presencia de colesterol en su estructura. Los azoles presentan un perfil de absorción variable, especialmente en el caso del itraconazol y del posaconazol, este último muy dependiente de múltiples factores. En el caso del voriconazol puede existir variabilidad a este respecto, lo que obliga a considerar la posibilidad de realizar una monitorización de las concentraciones plasmáticas.
El objetivo de este artículo es revisar algunos de los aspectos más relevantes de la farmacología de los antifúngicos utilizados en la profilaxis y el tratamiento de la infección aspergilar. Por ello se incluirán los aspectos más relevantes de algunos de los azoles que suelen prescribirse en esta infección (itraconazol, posaconazol y voriconazol) y de las formulaciones de anfotericina B.
The treatment of invasive aspergillosis requires the use of drugs that characteristically have complex pharmacokinetic properties, the knowledge of which is essential to achieve maximum efficacy with minimal risk to the patient. The lipid-based amphotericin B formulations vary significantly in their pharmacokinetic behaviour, with very high plasma concentrations of the liposomal form, probably related to the presence of cholesterol in their structure. Azoles have a variable absorption profile, particularly in the case of itraconazole and posaconazole, with the latter very dependent on multiple factors. This may also lead to variations in voriconazole, which requires considering the possibility of monitoring plasma concentrations.
The aim of this article is to review some of the most relevant aspects of the pharmacology of the antifungals used in the prophylaxis and treatment of the Aspergillus infection. For this reason, it includes the most relevant features of some of the azoles normally prescribed in this infection (itraconazole, posaconazole and voriconazole) and the amphotericin B formulations.
La estructura de los lípidos de las formulaciones de anfotericina B es diferente; el complejo lipídico contiene l-α-dimiristoilfosfatililcolina y l-α-dimiristoilfosfatidilglicerol, y la forma liposómica fosfatidilcolina, colesterol y diestearoilfosfatidilglicerol. Esta diferencia justifica un tamaño muy grande en el caso del complejo lipídico (1,6-11μm), frente al del liposoma (55-75nm). Además, la presencia de colesterol en el liposoma es responsable de proporcionar gran estabilidad a esta macroestructura. El complejo lipídico se forma en ausencia de colesterol y en él participa el l-α-dimiristoilfosfatililcolina, lo que explica que la estabilidad de esta macroestructura sea dependiente de la temperatura, ya que este fosfolípido tiene una temperatura límite de transición de 23°C (vs. una temperatura de 55°C que muestra la fuerte estabilidad de la formulación liposómica), lo que considerando la temperatura corporal sería una fuente potencial de inestabilidad del complejo lipídico1.
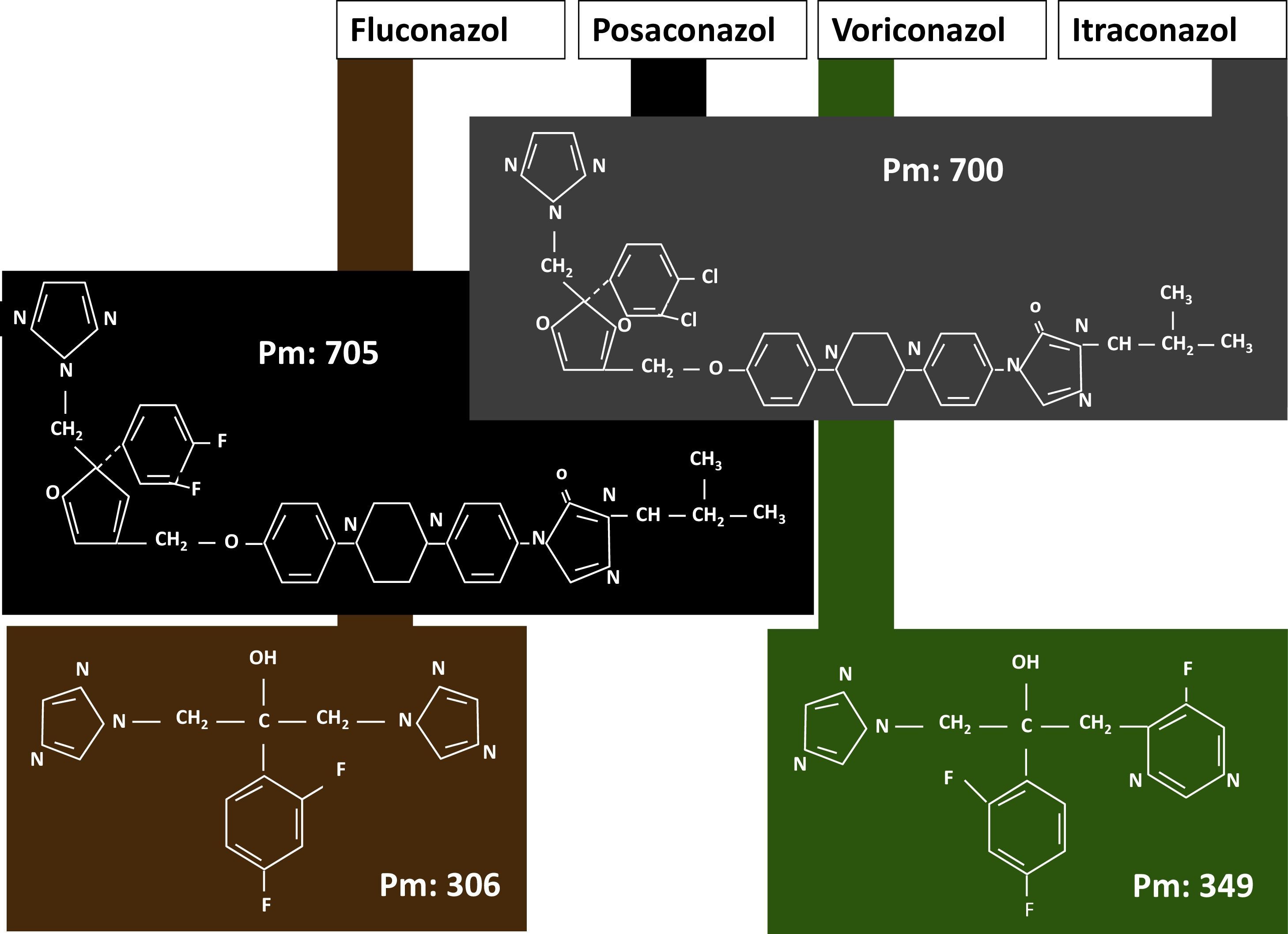
Los azoles, atendiendo a su estructura química (fig. 1), pueden agruparse en dos subgrupos: el formado por el itraconazol y el posaconazol, que presentan una estructura central similar con la única diferencia del tipo de halógeno, y un segundo subgrupo formado por el voriconazol, que es una fluoropirimidina triazólica. Esta diferencia es muy evidente en cuanto a los pesos moleculares, de 349Da para el voriconazol, y más de 700Da para el itraconazol y el posaconazol.
Farmacocinética
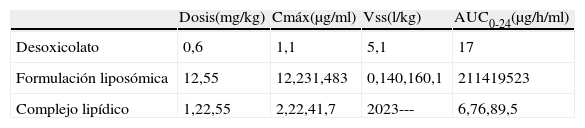
La absorción de la anfotericina B tras la administración oral es inexistente. Tras la administración intravenosa de las diferentes formulaciones de anfotericina B (tabla 1) se puede comprobar que la formulación liposómica alcanza concentraciones plasmáticas mucho más elevadas que las otras dos formulaciones y, al mismo tiempo, las concentraciones que se consiguen con estas dos son parecidas entre sí. El volumen de distribución de la formulación liposómica es muy inferior al de las otras dos formulaciones, mientras que el área bajo la curva (AUC) es mucho más elevada3,12,56.
El voriconazol presenta una biodisponibilidad elevada (80-90%) cuando se administra por vía oral en voluntarios83, pero esta biodisponibilidad es inferior en otras circunstancias de administración, como ocurre con el posaconazol y el itraconazol, cuya biodisponibilidad se sitúa entre el 40 y el 60%67,71.
La biodisponibilidad del itraconazol mejora hasta un 30-40% cuando se administra con betaciclodextrina50.
El posaconazol tiene una característica muy peculiar, difícil de encontrar en el resto de la farmacología, ya que su biodisponibilidad no solo no se relaciona de forma directa con la dosis, sino que es menor conforme mayor es la dosis administrada. Al parecer este problema está producido por dificultades en la solubilidad. Estas características han de ser tenidas en cuenta a la hora de dosificar este fármaco, ya que intentar mejorar la comodidad de la administración reduciendo el número de dosis se asocia con una reducción notable de la biodisponibilidad38.
La administración de itraconazol asociado a famotidina, ranitidina o antiácidos orales reduce la biodisponibilidad hasta un 50%58,65,68, circunstancia que parece aliviarse en parte si, al mismo tiempo, se ingiere una bebida de características ácidas o con vitamina C o vinagre7,55,78. La formulación en solución no presenta estos problemas57.
La elevación del pH gástrico inducida por los inhibidores de la bomba de protones (IBP) reduce de forma muy importante la concentración máxima y el AUC de posaconazol, mientras que la administración junto con un suplemento nutricional aumenta ambos valores62. Es posible que los antihistamínicos H2 produzcan menor reducción de la biodisponibilidad de posaconazol que los IBP, aunque el efecto tiene como consecuencia concentraciones de posaconazol inferiores a las consideradas como terapéuticas89.
La administración asociada de IBP (concretamente, de omeprazol) y voriconazol produce un ligero aumento de la concentración máxima y del AUC como consecuencia de la reducción de la actividad del sistema micosomal inducido por el IBP111. Esta circunstancia ha sido utilizada para mejorar la biodisponibilidad del antifúngico18. Los antihistamínicos H2 no producen efectos significativos sobre su biodisponibilidad85.
Los alimentos producen un aumento de la biodisponibilidad del itraconazol cuando es administrado en forma de cápsulas9. Este efecto se invierte cuando se administra en forma de solución, ya que en este caso la biodisponibilidad aumenta cuando se ingiere en ayunas104. La ingesta de posaconazol conjuntamente con comida grasa se acompaña de un aumento del AUC de hasta 4 veces en comparación con la ingesta en ayunas. La administración junto con una comida de menor contenido en grasa produce un aumento menor30. La administración con bebida de cola o con un suplemento nutricional (Boots plus®) mejoró la biodisponibilidad del posaconazol, generando valores de AUC superiores a los observados cuando se administró con agua62,92. La mejoría de la absorción del posaconazol producida por la bebida de cola se atribuye a una mejoría de la solubilidad. La ingesta de voriconazol junto con alimentos lleva a una reducción moderada, en el límite de la significación clínica, de la biodisponibilidad de este antifúngico. Por ello, cuando sea preciso optimizar la forma de administración debe preferirse la administración en ayunas84.
DistribuciónAnfotericina BLa biodisponibilidad de la anfotericina B cuando se administra en forma liposómica es mucho más elevada, con valores de volumen de distribución muy reducidos y aclaramiento también muy reducido. Se señala como hipótesis que, probablemente, la capacidad del liposoma para ser secuestrado de la circulación por las células y, tras ello, ser transportado a compartimentos profundos, podría justificar las mencionadas diferencias.
En un estudio realizado en voluntarios sanos para evaluar el modo en el que se encuentra la anfotericina B en la sangre cuando se administran dosis intravenosas de 2mg/kg de la formulación lipídica durante 2h, se comprobó que la mayor parte de la anfotericina B plasmática se encontraba dentro de los liposomas13. La proporción de fármaco ubicado fuera del liposoma es escasa y, además, se encuentra en su gran mayoría fijado a proteínas plasmáticas, concretamente a lipoproteínas. De hecho, la fracción libre, expresada en este caso como concentración plasmática de anfotericina B libre, no supera los 0,01μg/ml en ningún momento tras la administración del fármaco, aunque se mantienen estos valores hasta 168h después de la administración. Se ha señalado que el porcentaje de anfotericina B libre en ningún caso es superior al 5% de la concentración total del fármaco presente en el plasma.
La escasa proporción de fármaco libre no limita la difusión de la anfotericina B liposómica a los tejidos, tal y como se ha demostrado en varios estudios realizados en animales de experimentación11,44 y en el ser humano47,109. En general, la concentración es elevada en la práctica totalidad de los tejidos, especialmente en los que sufren infección, exceptuando el parénquima renal, donde es muy reducida con la formulación liposómica. Este hecho se ha relacionado con la capacidad de la anfotericina B liposómica de inhibir la actividad de las proteínas de transferencia de lípidos, lo que tiene como consecuencia que las moléculas de este antifúngico que circulan fijadas a lipoproteínas de alta densidad (HDL) no puedan ser transferidas a lipoproteínas de baja densidad (LDL), cuyos receptores no son expresados en células renales110.
Con dosis de anfotericina B liposómica de 5mg/kg/día se consiguen concentraciones en el tejido del sistema nervioso central próximas a 2μg/g, frente a cifras hasta diez veces inferiores en el caso del complejo lipídico y en el de anfotericina B desoxicolato, en un modelo de de meningoencefalitis por Candida albicans en conejo43. Las concentraciones detectadas en el líquido cefalorraquídeo (LCR) de las todas las formulaciones de anfotericina B ensayadas eran inferiores a 0,1mg/l.
AzolesEl pequeño tamaño de las moléculas de los azoles, unido a su elevada lipofilia, supone que tengan un volumen de distribución elevado que se va a manifestar por la presencia de concentraciones plasmáticas reducidas e intracelulares elevadas. Esta circunstancia es especialmente llamativa en los casos del itraconazol y el posaconazol por su mayor lipofilia, y explica otra de las características de estos dos fármacos: la elevada proporción de fijación a proteínas con que circulan en plasma, que supone fracciones libres del fármaco inferiores al 1%. Esta característica contrasta con unos porcentajes de fijación a proteínas del 58% para el voriconazol. Como expresión de estas características el volumen de distribución del voriconazol, aunque elevado (4-6l/kg), es inferior a los descritos para el itraconazol (11-14l/kg) y el posaconazol (343-1.341l)28
La penetración intracelular de estos fármacos está bien documentada. En el caso del itraconazol la captación por parte de los macrófagos se produce de forma rápida, con acumulación intracelular, mediante un proceso que parece no requerir consumo de energía82. También se ha descrito que la relación concentración intracelular/concentración extracelular de posaconazol en células de estirpe hematológica es elevada39.
Recientemente se ha publicado un estudio in vitro en el que se ha evaluado la captación intracelular por parte de células epiteliales de diversos antifúngicos. Los resultados mostraron la mayor capacidad de penetración del posaconazol frente a la casi inexistente del voriconazol22, lo que contrasta con los resultados descritos en otros estudios8.
El itraconazol y su metabolito activo (el hidroxitraconazol)27, el posaconazol26 y el voriconazol23,31 alcanzan concentraciones adecuadas en el líquido de lavado alveolar y en las células del epitelio bronquial. Existe una diferencia notable en los valores de concentración en el líquido del epitelio pulmonar, que son muy superiores en el caso del voriconazol, que se ha relacionado con su menor porcentaje de fijación a proteínas (58%) en comparación con aquel de los restantes antifúngicos88.
La disponibilidad de concentraciones activas de antifúngico en el LCR y en el tejido cerebral parece un requisito importante a la hora de tratar cualquiera de las infecciones fúngicas invasoras, puesto que no es infrecuente la presencia de hongos en el tejido cerebral o en el LCR. En este caso, el voriconazol presenta una difusión importante al LCR45,70 en comparación con los restantes azoles87,95 o la anfotericina B96, lo que termina de mostrarse en su eficacia clínica32,93,100,.
No existe información sobre la penetración del itraconazol o el posaconazol al ojo, mientras que el voriconazol ha mostrado su capacidad para alcanzar concentraciones activas en el humor vítreo y el humor acuoso que suponen el 38-40% y el 53% de las plasmáticas, respectivamente46.
EliminaciónAnfotericina BLa eliminación de la anfotericina B se produce con gran lentitud y por vías no bien aclaradas. Se ha descrito la presencia de anfotericina B en las heces y la orina en porcentajes inferiores al 5% a lo largo de las 168h (7 días) posteriores a la administración del fármaco en su formulación liposómica. En el caso de la formulación convencional estos valores se sitúan en el 20,6 y 42,5% respectivamente13. Se ha estimado una semivida de eliminación que supera las 120h (más de 5 días). Es probable, si consideramos su estabilidad y el transporte intracelular, que el antifúngico permanezca dentro del liposoma durante largos periodos de tiempo y que solo se libere del mismo en circunstancias muy concretas.
AzolesLos azoles se eliminan a través del metabolismo en el que intervienen de forma decisiva algunas de las isoenzimas del CYP450.
El itraconazol se transforma en tres metabolitos: el hidroxi-itraconazol, el keto-itraconazol y el N-desalquil-itraconazol, todos ellos provistos de capacidad para inhibir la actividad de la CYP3A4101. El posaconazol se elimina a través del metabolismo mediante reacciones de fase ii, transformándose en derivados glucuroconjugados por intervención de la UDP-glucuronosiltransferasa (UGT) fracción UGT1A4, que son eliminados a través de la orina61. La metabolización del voriconazol se produce a través de la N-oxidación de la fluoropirimidina, posterior hidroxilación de la fluoropirimidina, y su metilhidroxilación y posterior conjugación con ácido glucurónico40. En la metabolización del voriconazol intervienen las isoenzimas CYP2C9, CYP2C19 y CYP3A4. También se ha descrito la implicación de una monooxigenasa con contenido de flavina en la formación de N-óxido, cuya mayor actividad en niños podría relacionarse con la necesidad de utilizar dosis mayores de este fármaco112. La participación de la CYP2C19 en el metabolismo del voriconazol justifica la variabilidad relacionada con polimorfismos42,59, pero no toda la variabilidad descrita.
El itraconazol tiene un semivida de 34-48h La semivida terminal del voriconazol es mucho más corta, de 6,5-6,7h83, mientras que para el posaconazol se han descrito tiempos parecidos a los del itraconazol: 25-30h. Es por ello que la consecución del estado de equilibrio cuando no se utiliza una dosis de carga, como es el caso del posaconazol, y la persistencia de concentraciones plasmáticas capaces de producir efectos adversos e interacciones sean diferentes: uno a dos días para el voriconazol, y cinco a seis días para el posaconazol y el itraconazol.
InteraccionesLa anfotericina no está implicada en interacciones farmacocinéticas con otros fármacos. En cambio, es posible que algunos de los efectos adversos y, especialmente, la nefrotoxicidad y la hipopotasemia sean más frecuentes e importantes cuando se utiliza asociada a otros fármacos que producen efectos similares.
Los antifúngicos azólicos comparten mecanismo de acción, que consiste en la reducción de la síntesis del ergosterol por inhibición de una metilasa perteneciente a la dotación enzimática ligada al sistema CYP450 del hongo. La similitud estructural entre esta enzima y las presentes en el ser humano implica que los fármacos de este grupo establezcan interacciones, cuyo denominador común va a ser la capacidad del antifúngico para inhibir alguna de las isoenzimas del sistema CYP450 del huésped. Los azoles presentan algunas diferencias en su estructura química que les confiere comportamientos distintos en lo referente a su potencial inhibidor de las isoenzimas CYP450, aunque todos ellos presentan una actividad inhibitoria entre moderada y elevada75,77,106.
El voriconazol es un potente inhibidor de la CYP2C19, y un inhibidor moderado de la CYP2C9 y de la CYP3A56. El posaconazol y el itraconazol presentan una actividad inhibitoria potente de la CYP3A4 y, al mismo tiempo, de la glucoproteína P, proteína que participa en la absorción de muchos fármacos a través de su papel como bomba de expulsión desde la célula a la luz intestinal53.
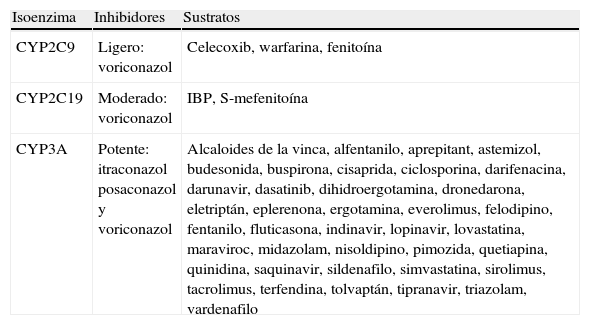
La combinación de un azol con cualquiera de los fármacos que son habitualmente el sustrato de la isoenzima inhibida produce una reducción de la velocidad de eliminación del fármaco, lo que se expresa en la reducción de su aclaramiento, que generará un aumento de la semivida de eliminación y la acumulación del fármaco, con el correspondiente riesgo de efectos adversos si la dosis no es reducida. La casuística existente es tan numerosa que resulta imposible incluirla en un texto ya demasiado largo, por ello se remite al lector a la literatura genérica del tema20,34,41,45,76,90,91,94,102. El conocimiento de los fármacos que son sustratos de las diferentes isoenzimas permite identificar con facilidad cuáles de ellos sufrirán interacciones con los distintos azoles53 (tabla 2).
Sustratos de los isoenzimas del CYP450 que son inhibidos por los azoles
| Isoenzima | Inhibidores | Sustratos |
| CYP2C9 | Ligero: voriconazol | Celecoxib, warfarina, fenitoína |
| CYP2C19 | Moderado: voriconazol | IBP, S-mefenitoína |
| CYP3A | Potente: itraconazol posaconazol y voriconazol | Alcaloides de la vinca, alfentanilo, aprepitant, astemizol, budesonida, buspirona, cisaprida, ciclosporina, darifenacina, darunavir, dasatinib, dihidroergotamina, dronedarona, eletriptán, eplerenona, ergotamina, everolimus, felodipino, fentanilo, fluticasona, indinavir, lopinavir, lovastatina, maraviroc, midazolam, nisoldipino, pimozida, quetiapina, quinidina, saquinavir, sildenafilo, simvastatina, sirolimus, tacrolimus, terfendina, tolvaptán, tipranavir, triazolam, vardenafilo |
Fármacos inductores (pueden aumentar la velocidad de eliminación de los azoles): fenitina, fenobarbital, carbamazepina, rifampicina.
Tal y como cabría esperar de un fármaco con estas características, la ausencia de eliminación renal y el elevado tamaño de la estructura que lo transporta hace que el impacto en la función renal sea mínimo, y tampoco se espera que el efecto de la hemodiálisis o el de cualquier otra técnica de depuración externa sea relevante4,10,14,15,107.
AzolesEl itraconazol no muestra en su farmacocinética cambios dependientes de la presencia de alteraciones de la función renal, por lo que no precisa ajuste de la dosis17. La formulación intravenosa contiene hidroxipropil-ß-ciclodextrina, solubilizante que se elimina a través de la filtración glomerular, que potencialmente podría producir alteraciones renales114. Por ello, está contraindicado en pacientes con insuficiencia renal grave (ClCr<30ml/min). Si se sospecha alteración de la función renal deberá considerarse el cambio a la formulación oral en cápsulas, evitando la solución ya que también contiene ciclodextrina que potencialmente podría acumularse. El posaconazol no sufre modificaciones cuando existe insuficiencia renal, y es apenas eliminado a través de hemodiálisis convencional29.
La administración de voriconazol en pacientes con insuficiencia renal no cursa con alteraciones de la farmacocinética del antifúngico2,74, aunque en el caso de la formulación intravenosa debe considerarse la presencia de ciclodextrina, si bien esta formulación no se ha asociado con toxicidad específica2,5,74.
El uso de técnicas de depuración externa, como la hemodiafiltración venovenosa continua, no elimina el voriconazol86, mientras que la ciclodextrina es totalmente eliminada a través de cualquiera de estas técnicas69. El voriconazol se elimina en escasa cantidad (menos del 1%) a través de la diálisis peritoneal81; tampoco el uso de técnicas de plasmaféresis elimina cantidades significativas de este antifúngico98. El uso de circulación extracorpórea puede modificar las concentraciones del voriconazol, por lo que se recomienda monitorizar sus concentraciones plasmáticas para un mejor ajuste.
No existe información específica del impacto de una función hepática alterada sobre la farmacocinética del itraconazol. Su utilización, por tanto, debe realizarse con las máximas precauciones.
El posaconazol puede experimentar un aumento de su concentración plasmática expresado como un incremento medio del 36% del AUC cuando existe alteración de la función hepática, por lo que se recomienda precaución con este antifúngico72.
En el caso del voriconazol se recomienda utilizar las dosis de carga normales, pero reducir a la mitad la dosis de mantenimiento en aquellos pacientes con cirrosis hepática de leve a moderada (Child Pugh A y B). No se ha estudiado la farmacocinética en pacientes con cirrosis hepática crónica grave (Child-Pugh C), aunque se han publicado casos que señalan que el fármaco se acumula (Fuhmann).
Se ha recomendado administrar una dosis de itraconazol de 2,5mg/kg/12h en niños a partir de 5 años1. El posaconazol se ha evaluado en sujetos jóvenes (edades comprendidas entre los 8 y los 17 años), en los que se ha comprobado la ausencia de diferencias en los parámetros farmacocinéticos en comparación con los adultos63. No existe información de niños de menor edad. La farmacocinética del voriconazol en niños se caracteriza por una elevada variabilidad, por lo que se recomienda monitorizar sus concentraciones plasmáticas10,99. Además, se ha descrito una biodisponibilidad oral inferior a la de los pacientes adultos, aclaramiento mas rápido y AUC inferior, que en su conjunto exigen dosis más elevadas y que en el caso de la formulación intravenosa se sitúa en 7-8mg/kg73,108. Se ha señalado que la mayor actividad en niños de la CYP2C19 y de una flavinmonooxigenasa (FMO3) sería la responsable de la mayor variabilidad farmacocinética y del aumento del aclaramiento del voriconazol en este grupo de edad113.
Recientemente se ha descrito que en pacientes con obesidad (IMC>35kg/m2) los valores de concentración mínima de voriconazol, administrado a dosis de 4mg/kg cada 12h, eran más elevados que los valores de pacientes sin obesidad. El porcentaje de pacientes que presentaban concentraciones supraterapéuticas (concentración mínima>5,5mg/l) era del 67%, frente al 17% en los sujetos sin obesidad. Por ello, los autores recomiendan ajustar la dosis al peso ideal o al peso real ajustado60. Se han descrito resultados similares en un estudio realizado en ocho voluntarios con obesidad de grado ii o superior, que presentaron valores de AUC semejantes a los de los pacientes sin sobrepeso cuando recibieron 200 o 300mg de voriconazol79.
No existe información específica sobre la dosificación del posaconazol o del itraconazol en el paciente obeso.
Monitorización de las concentraciones plasmáticasLa variabilidad de las concentraciones y la posibilidad de que algunos de los cuadros de toxicidad pudieran relacionarse con concentraciones demasiado elevadas, son los dos argumentos más utilizados para proponer el seguimiento de las concentraciones plasmáticas de los azoles que ya se propusieron con el itraconazol (con valores de concentración mínima de 250-500ng/ml)24,34,66.
En los últimos años se han acumulado evidencias acerca de la variabilidad de la farmacocinética del posaconazol. Las concentraciones reducidas de este fármaco se han relacionado con diversas circunstancias clínicas asociadas al paciente o a su tratamiento: diarrea, tratamiento asociado con IBP o antagonistas H2, peso corporal, edad, mucositis, náuseas, vómitos, administración de quimioterapia, enfermedad injerto contra huésped, ingesta reducida de alimentos o el uso de inductores del metabolismo, como fenitoína o rifampicina. Estos hallazgos explican la necesidad de monitorizar las concentraciones plasmáticas del posaconazol de forma rutinaria. Se han propuesto valores de concentración mínima de 0,5-1,5mg/l para lograr la cura de los procesos infecciosos, y de 0,5mg/l para la eficacia en la profilaxis6,16,35–37,48,49,52,103,105.
El voriconazol ha sido objeto de numerosas iniciativas dirigidas a su concentración plasmática y, especialmente, a los valores de concentración mínima, por su variable farmacocinética en relación al CYP2C19, la gravedad de la enfermedad para la que es utilizado de forma habitual y la posibilidad de que pueda existir un rango terapéutico. Se considera que su concentración debe situarse en valores iguales o superiores a 1mg/l para asegurar la eficacia del tratamiento, e inferiores a 6mg/l para evitar efectos adversos19,21,25,33,51,97. Para algunos autores solo estaría indicada en algunas circunstancias como pérdida de eficacia y evidencia de efectos adversos64.
La monitorización de la concentración de voriconazol ha sido de utilidad para proponer una modificación de la dosis del fármaco, basada en el análisis de regresión multivariado de los resultados obtenidos en pacientes con micosis invasora. La amplia variabilidad observada en la concentración y el aumento notable de la probabilidad de alcanzar concentraciones terapéuticas y no tóxicas del fármaco cuando se utilizaban dosis de 300mg diarios, en lugar de los 200mg diarios habituales, llevaron a la conclusión de que la dosis más idónea puede ser 300mg/12h80.
Conflicto de interesesJ. R. Azanza: conferencias, docencia y publicaciones con: Pfizer, Astellas, Gilead y MSD. El resto de autores declara no tener ningún conflicto de intereses.

