





The prevalence of opportunistic yeast infections has increased in recent decades as the result of an increasing immunocompromised patient population.
AimsTo evaluate ribosomal RNA (rRNA) gene sequence to identify medically important yeast species, to investigate the performance of both the rRNA gene internal transcribed spacer (ITS) and D1/D2 region in identifying clinically relevant yeasts, and to compare these results with those of a standard phenotypic method.
MethodsBoth regions from 50 yeast strains, comprising 45 clinical isolates and 5 reference strains, were amplified using PCR and then sequenced. The sequences were compared to reference data available from the GenBank database of the National Center for Biotechnology Information using the BLASTn tool.
ResultsUsing ID32C, 88% (44/50) of all strains were identified accurately at the species level, although 6% were misidentified; two Candida eremophila isolates were identified as Candida glabrata and Candida tropicalis, and one Saprochaete clavata isolate was identified as Saprochaete capitata. Two of the four isolates identified by phenotypic methods as Trichosporon asahii were defined so by analyzing the ITS region, but the remaining two were not distinguishable from closely related species. Based on the D1/D2 region, these four isolates had 100% sequence identity with T. asahii, Trichosporon japonicum, and Trichosporon asteroides. The isolate identified as Trichosporon inkin using ID32C could not be distinguished from Trichosporon ovoides by analyzing the ITS and D1/D2 regions.
ConclusionsIdentifying medically important yeasts by sequencing the ITS and D1/D2 region is a rapid and reliable alternative to conventional identification methods. For a diagnostic algorithm, we suggest a two-step procedure integrating conventional methods (e.g. microscopic morphology on corn meal agar with Tween® 80 and API ID32C®) and sequence analysis of the ITS and D1/D2 region.
La prevalencia de infecciones oportunistas por levaduras ha aumentado en las últimas décadas como resultado de una población de pacientes inmunocomprometidos cada vez mayor.
ObjetivosEvaluar la secuencia del gen del ARN ribosomal (ARNr) para identificar especies de levaduras médicamente importantes, investigar el rendimiento del espaciador transcrito interno del gen ARNr (ITS) y las regiones D1/D2 en la identificación de levaduras clínicamente relevantes, y comparar estos resultados con los de un método fenotípico estándar.
MétodosAmbas regiones del ARNr de 50 cepas de levaduras con 45 aislamientos clínicos y 5 cepas de referencia se amplificaron mediante PCR y posteriormente se secuenciaron. Las secuencias se compararon con los datos de referencia disponibles en la base de datos GenBank® del Centro Nacional de Información Biotecnológica mediante la herramienta BLASTn.
ResultadosMediante el método ID32C el 88% (44/50) de todas las cepas se identificaron con precisión y el 6% se identificaron erróneamente; dos aislamientos de Candida eremophila fueron identificados como Candida glabrata y Candida tropicalis, y un aislamiento de Saprochaete clavata fue identificado como Saprochaete capitata. Dos de los cuatro aislamientos identificados por métodos fenotípicos como Trichosporon asahii se catalogaron así al analizar la región ITS, pero las dos restantes no se distinguían de las especies estrechamente relacionadas. En base a la secuencia de la región D1/D2, estos cuatro aislamientos se identificaron, con un 100% de similitud, como T. asahii, Trichosporon japonicum y Trichosporon asteroides. El aislamiento identificado como Trichosporon inkin mediante ID32C no se pudo distinguir de Trichosporon ovoides al analizar las regiones ITS y D1/D2.
ConclusionesLa identificación de levaduras de interés médico mediante la secuenciación de las regiones ITS y D1/D2 es una alternativa rápida y confiable a los métodos de identificación convencionales. Para un algoritmo de diagnóstico sugerimos un procedimiento de dos pasos que integre métodos convencionales (morfología microscópica en agar de harina de maíz con Tween® 80 y API ID32C®) y análisis de la secuencia de las regiones ITS y D1/D2.
The prevalence of fungal infections has increased in the last two decades in parallel with the increasing number of immunosuppressed patients and advances in medical treatments.19 Facilitating factors, such as increasingly frequent surgical interventions, including tissue and organ transplantation, the use of immunosuppressive agents, and central venous catheter and mechanical ventilation applications, have increased the morbidity and mortality rates of opportunistic fungal infections.21,23
Although Candida albicans is still the most common cause of opportunistic fungal infections, over 150 fungal species belonging to Candida and other genera may cause human infections.2 The common use of antifungal agents for prophylaxis has been suggested to contribute to the emergence of naturally resistant strains as important pathogens by shifting the distribution of species; in particular, non-C. albicans species are increasingly isolated as a result of azole-derivative antifungal use.1,2 In recent years, infections caused by less common yeast-like fungi, such as Cryptococcus, Trichosporon, Saprochaete, Pichia, Rhodotorula, and Saccharomyces, have increased.14,16,18,31
The early, rapid, and accurate identification of yeasts is important for prognostic, epidemiological, and therapeutic reasons.18 Distinguishing yeasts isolated from clinical specimens at the species level is important to determine disease etiology, identify new agents, select the appropriate antifungal therapy, and prevent the spread of the agent to susceptible patient populations.11,21 Conventional methods to identify yeasts at the species level are the germ tube test, the carbohydrate assimilation–fermentation test, and the use of corn meal agar with Tween® 80. Existing commercial identification systems that traditionally identify pathogenic yeasts based on morphological and biochemical characteristics are time consuming, have limited databases, and are designed to identify only the more common pathogens.2,17 Therefore, molecular approaches have been developed as an alternative to conventional methods to ensure a more rapid and accurate identification of fungi. Among these molecular approaches, the sequencing of ribosomal genes has emerged as a valuable diagnostic tool that allows the rapid detection and identification of fungi.20
The most widely used ribosomal target for sequence analysis is the internal transcribed spacer (ITS) region. Although the ITS region sequence may identify many fungi at the species level based on sequence variability, it alone cannot accurately identify certain fungi. To resolve this, sequencing the second variable site in the ribosomal DNA (rDNA) cluster, referred to as the D1/D2 region, is recommended.9,12,21,27 In recent years, matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI–TOF MS) has emerged in clinical mycology laboratories as an alternative for fungal identification.26
The aims of the present study were to evaluate DNA sequencing analysis using the ribosomal RNA (rRNA) gene ITS and D1/D2 regions to identify medically important yeast isolates at the species level, and to compare these results with those of conventional methods.
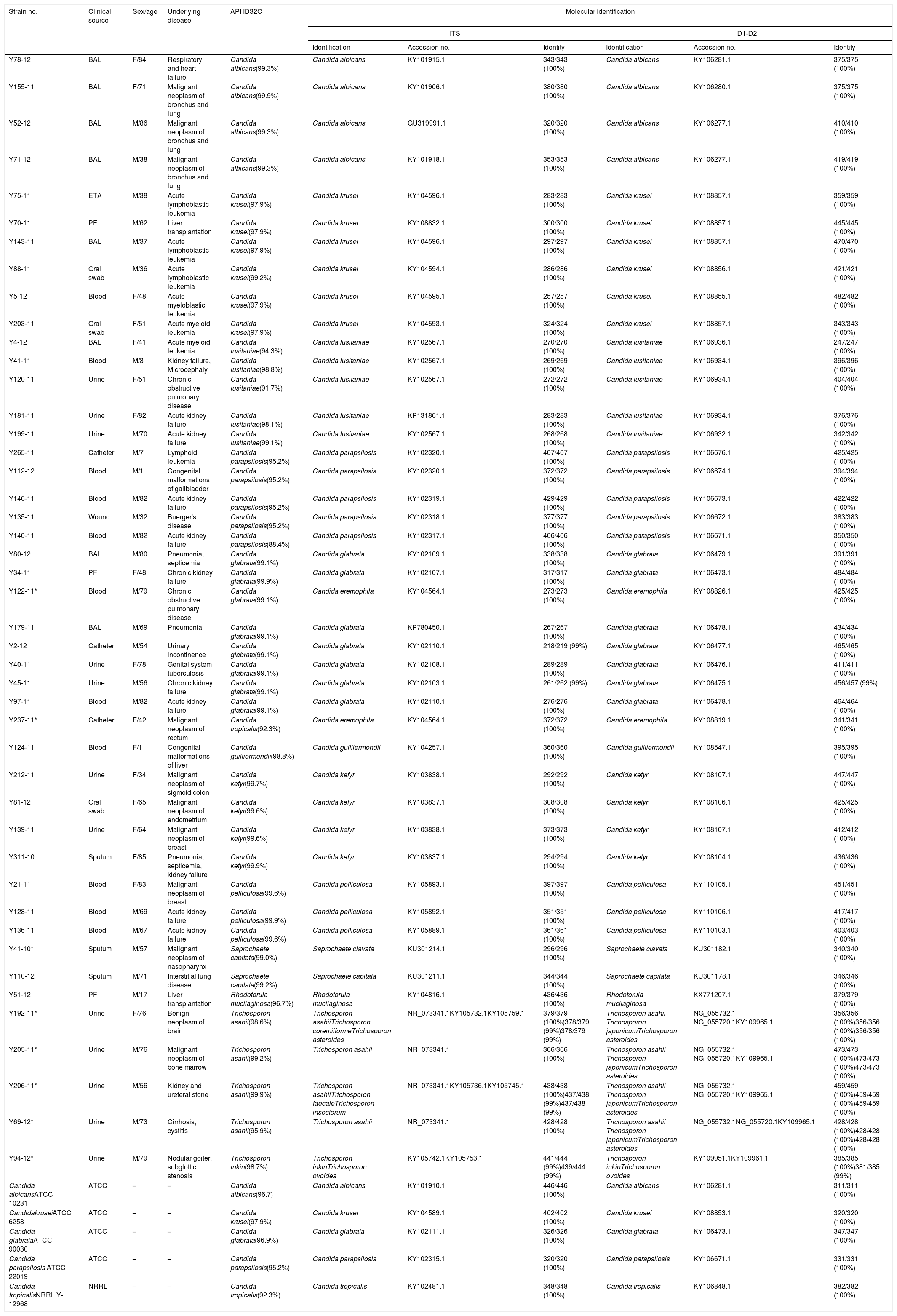
Materials and methodsReference strains and isolatesThis study was approved by the Gazi University Faculty of Medicine Ethical Committee. Fifty yeast isolates were used, of which five were reference strains (C. albicans ATCC 10231, Candida krusei ATCC 6258, Candida glabrata ATCC 90030, Candida parapsilosis ATCC 22019, and Candida tropicalis NRRL Y-12968). The remaining 45 clinical isolates were selected from the depository of fungal strains from the Gazi University Medical Mycology Laboratory, based on their phenotypic identification. The isolate selection criteria were as follows: (1) those most frequently recovered in our laboratory, or (2) those of medical importance that are infrequently recovered. The isolates were obtained from a variety of clinical sources: 12 urine, 11 blood, 8 bronchoalveolar lavage, 3 catheter, 3 oral swab, 3 peritoneal fluid, 3 sputum, 1 endotracheal aspirate, and 1 wound (Table 1).
Phenotypic and genotypic identification of the yeast isolates analyzed in this study.
| Strain no. | Clinical source | Sex/age | Underlying disease | API ID32C | Molecular identification | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| ITS | D1-D2 | |||||||||
| Identification | Accession no. | Identity | Identification | Accession no. | Identity | |||||
| Y78-12 | BAL | F/84 | Respiratory and heart failure | Candida albicans(99.3%) | Candida albicans | KY101915.1 | 343/343 (100%) | Candida albicans | KY106281.1 | 375/375 (100%) |
| Y155-11 | BAL | F/71 | Malignant neoplasm of bronchus and lung | Candida albicans(99.9%) | Candida albicans | KY101906.1 | 380/380 (100%) | Candida albicans | KY106280.1 | 375/375 (100%) |
| Y52-12 | BAL | M/86 | Malignant neoplasm of bronchus and lung | Candida albicans(99.3%) | Candida albicans | GU319991.1 | 320/320 (100%) | Candida albicans | KY106277.1 | 410/410 (100%) |
| Y71-12 | BAL | M/38 | Malignant neoplasm of bronchus and lung | Candida albicans(99.3%) | Candida albicans | KY101918.1 | 353/353 (100%) | Candida albicans | KY106277.1 | 419/419 (100%) |
| Y75-11 | ETA | M/38 | Acute lymphoblastic leukemia | Candida krusei(97.9%) | Candida krusei | KY104596.1 | 283/283 (100%) | Candida krusei | KY108857.1 | 359/359 (100%) |
| Y70-11 | PF | M/62 | Liver transplantation | Candida krusei(97.9%) | Candida krusei | KY108832.1 | 300/300 (100%) | Candida krusei | KY108857.1 | 445/445 (100%) |
| Y143-11 | BAL | M/37 | Acute lymphoblastic leukemia | Candida krusei(97.9%) | Candida krusei | KY104596.1 | 297/297 (100%) | Candida krusei | KY108857.1 | 470/470 (100%) |
| Y88-11 | Oral swab | M/36 | Acute lymphoblastic leukemia | Candida krusei(99.2%) | Candida krusei | KY104594.1 | 286/286 (100%) | Candida krusei | KY108856.1 | 421/421 (100%) |
| Y5-12 | Blood | F/48 | Acute myeloblastic leukemia | Candida krusei(97.9%) | Candida krusei | KY104595.1 | 257/257 (100%) | Candida krusei | KY108855.1 | 482/482 (100%) |
| Y203-11 | Oral swab | F/51 | Acute myeloid leukemia | Candida krusei(97.9%) | Candida krusei | KY104593.1 | 324/324 (100%) | Candida krusei | KY108857.1 | 343/343 (100%) |
| Y4-12 | BAL | F/41 | Acute myeloid leukemia | Candida lusitaniae(94.3%) | Candida lusitaniae | KY102567.1 | 270/270 (100%) | Candida lusitaniae | KY106936.1 | 247/247 (100%) |
| Y41-11 | Blood | M/3 | Kidney failure, Microcephaly | Candida lusitaniae(98.8%) | Candida lusitaniae | KY102567.1 | 269/269 (100%) | Candida lusitaniae | KY106934.1 | 396/396 (100%) |
| Y120-11 | Urine | F/51 | Chronic obstructive pulmonary disease | Candida lusitaniae(91.7%) | Candida lusitaniae | KY102567.1 | 272/272 (100%) | Candida lusitaniae | KY106934.1 | 404/404 (100%) |
| Y181-11 | Urine | F/82 | Acute kidney failure | Candida lusitaniae(98.1%) | Candida lusitaniae | KP131861.1 | 283/283 (100%) | Candida lusitaniae | KY106934.1 | 376/376 (100%) |
| Y199-11 | Urine | M/70 | Acute kidney failure | Candida lusitaniae(99.1%) | Candida lusitaniae | KY102567.1 | 268/268 (100%) | Candida lusitaniae | KY106932.1 | 342/342 (100%) |
| Y265-11 | Catheter | M/7 | Lymphoid leukemia | Candida parapsilosis(95.2%) | Candida parapsilosis | KY102320.1 | 407/407 (100%) | Candida parapsilosis | KY106676.1 | 425/425 (100%) |
| Y112-12 | Blood | M/1 | Congenital malformations of gallbladder | Candida parapsilosis(95.2%) | Candida parapsilosis | KY102320.1 | 372/372 (100%) | Candida parapsilosis | KY106674.1 | 394/394 (100%) |
| Y146-11 | Blood | M/82 | Acute kidney failure | Candida parapsilosis(95.2%) | Candida parapsilosis | KY102319.1 | 429/429 (100%) | Candida parapsilosis | KY106673.1 | 422/422 (100%) |
| Y135-11 | Wound | M/32 | Buerger's disease | Candida parapsilosis(95.2%) | Candida parapsilosis | KY102318.1 | 377/377 (100%) | Candida parapsilosis | KY106672.1 | 383/383 (100%) |
| Y140-11 | Blood | M/82 | Acute kidney failure | Candida parapsilosis(88.4%) | Candida parapsilosis | KY102317.1 | 406/406 (100%) | Candida parapsilosis | KY106671.1 | 350/350 (100%) |
| Y80-12 | BAL | M/80 | Pneumonia, septicemia | Candida glabrata(99.1%) | Candida glabrata | KY102109.1 | 338/338 (100%) | Candida glabrata | KY106479.1 | 391/391 (100%) |
| Y34-11 | PF | F/48 | Chronic kidney failure | Candida glabrata(99.9%) | Candida glabrata | KY102107.1 | 317/317 (100%) | Candida glabrata | KY106473.1 | 484/484 (100%) |
| Y122-11* | Blood | M/79 | Chronic obstructive pulmonary disease | Candida glabrata(99.1%) | Candida eremophila | KY104564.1 | 273/273 (100%) | Candida eremophila | KY108826.1 | 425/425 (100%) |
| Y179-11 | BAL | M/69 | Pneumonia | Candida glabrata(99.1%) | Candida glabrata | KP780450.1 | 267/267 (100%) | Candida glabrata | KY106478.1 | 434/434 (100%) |
| Y2-12 | Catheter | M/54 | Urinary incontinence | Candida glabrata(99.1%) | Candida glabrata | KY102110.1 | 218/219 (99%) | Candida glabrata | KY106477.1 | 465/465 (100%) |
| Y40-11 | Urine | F/78 | Genital system tuberculosis | Candida glabrata(99.1%) | Candida glabrata | KY102108.1 | 289/289 (100%) | Candida glabrata | KY106476.1 | 411/411 (100%) |
| Y45-11 | Urine | M/56 | Chronic kidney failure | Candida glabrata(99.1%) | Candida glabrata | KY102103.1 | 261/262 (99%) | Candida glabrata | KY106475.1 | 456/457 (99%) |
| Y97-11 | Blood | M/82 | Acute kidney failure | Candida glabrata(99.1%) | Candida glabrata | KY102110.1 | 276/276 (100%) | Candida glabrata | KY106478.1 | 464/464 (100%) |
| Y237-11* | Catheter | F/42 | Malignant neoplasm of rectum | Candida tropicalis(92.3%) | Candida eremophila | KY104564.1 | 372/372 (100%) | Candida eremophila | KY108819.1 | 341/341 (100%) |
| Y124-11 | Blood | F/1 | Congenital malformations of liver | Candida guilliermondii(98.8%) | Candida guilliermondii | KY104257.1 | 360/360 (100%) | Candida guilliermondii | KY108547.1 | 395/395 (100%) |
| Y212-11 | Urine | F/34 | Malignant neoplasm of sigmoid colon | Candida kefyr(99.7%) | Candida kefyr | KY103838.1 | 292/292 (100%) | Candida kefyr | KY108107.1 | 447/447 (100%) |
| Y81-12 | Oral swab | F/65 | Malignant neoplasm of endometrium | Candida kefyr(99.6%) | Candida kefyr | KY103837.1 | 308/308 (100%) | Candida kefyr | KY108106.1 | 425/425 (100%) |
| Y139-11 | Urine | F/64 | Malignant neoplasm of breast | Candida kefyr(99.6%) | Candida kefyr | KY103838.1 | 373/373 (100%) | Candida kefyr | KY108107.1 | 412/412 (100%) |
| Y311-10 | Sputum | F/85 | Pneumonia, septicemia, kidney failure | Candida kefyr(99.9%) | Candida kefyr | KY103837.1 | 294/294 (100%) | Candida kefyr | KY108104.1 | 436/436 (100%) |
| Y21-11 | Blood | F/83 | Malignant neoplasm of breast | Candida pelliculosa(99.6%) | Candida pelliculosa | KY105893.1 | 397/397 (100%) | Candida pelliculosa | KY110105.1 | 451/451 (100%) |
| Y128-11 | Blood | M/69 | Acute kidney failure | Candida pelliculosa(99.9%) | Candida pelliculosa | KY105892.1 | 351/351 (100%) | Candida pelliculosa | KY110106.1 | 417/417 (100%) |
| Y136-11 | Blood | M/67 | Acute kidney failure | Candida pelliculosa(99.6%) | Candida pelliculosa | KY105889.1 | 361/361 (100%) | Candida pelliculosa | KY110103.1 | 403/403 (100%) |
| Y41-10* | Sputum | M/57 | Malignant neoplasm of nasopharynx | Saprochaete capitata(99.0%) | Saprochaete clavata | KU301214.1 | 296/296 (100%) | Saprochaete clavata | KU301182.1 | 340/340 (100%) |
| Y110-12 | Sputum | M/71 | Interstitial lung disease | Saprochaete capitata(99.2%) | Saprochaete capitata | KU301211.1 | 344/344 (100%) | Saprochaete capitata | KU301178.1 | 346/346 (100%) |
| Y51-12 | PF | M/17 | Liver transplantation | Rhodotorula mucilaginosa(96.7%) | Rhodotorula mucilaginosa | KY104816.1 | 436/436 (100%) | Rhodotorula mucilaginosa | KX771207.1 | 379/379 (100%) |
| Y192-11* | Urine | F/76 | Benign neoplasm of brain | Trichosporon asahii(98.6%) | Trichosporon asahiiTrichosporon coremiiformeTrichosporon asteroides | NR_073341.1KY105732.1KY105759.1 | 379/379 (100%)378/379 (99%)378/379 (99%) | Trichosporon asahii Trichosporon japonicumTrichosporon asteroides | NG_055732.1 NG_055720.1KY109965.1 | 356/356 (100%)356/356 (100%)356/356 (100%) |
| Y205-11* | Urine | M/76 | Malignant neoplasm of bone marrow | Trichosporon asahii(99.2%) | Trichosporon asahii | NR_073341.1 | 366/366 (100%) | Trichosporon asahii Trichosporon japonicumTrichosporon asteroides | NG_055732.1 NG_055720.1KY109965.1 | 473/473 (100%)473/473 (100%)473/473 (100%) |
| Y206-11* | Urine | M/56 | Kidney and ureteral stone | Trichosporon asahii(99.9%) | Trichosporon asahiiTrichosporon faecaleTrichosporon insectorum | NR_073341.1KY105736.1KY105745.1 | 438/438 (100%)437/438 (99%)437/438 (99%) | Trichosporon asahii Trichosporon japonicumTrichosporon asteroides | NG_055732.1 NG_055720.1KY109965.1 | 459/459 (100%)459/459 (100%)459/459 (100%) |
| Y69-12* | Urine | M/73 | Cirrhosis, cystitis | Trichosporon asahii(95.9%) | Trichosporon asahii | NR_073341.1 | 428/428 (100%) | Trichosporon asahii Trichosporon japonicumTrichosporon asteroides | NG_055732.1NG_055720.1KY109965.1 | 428/428 (100%)428/428 (100%)428/428 (100%) |
| Y94-12* | Urine | M/79 | Nodular goiter, subglottic stenosis | Trichosporon inkin(98.7%) | Trichosporon inkinTrichosporon ovoides | KY105742.1KY105753.1 | 441/444 (99%)439/444 (99%) | Trichosporon inkinTrichosporon ovoides | KY109951.1KY109961.1 | 385/385 (100%)381/385 (99%) |
| Candida albicansATCC 10231 | ATCC | – | – | Candida albicans(96.7) | Candida albicans | KY101910.1 | 446/446 (100%) | Candida albicans | KY106281.1 | 311/311 (100%) |
| CandidakruseiATCC 6258 | ATCC | – | – | Candida krusei(97.9%) | Candida krusei | KY104589.1 | 402/402 (100%) | Candida krusei | KY108853.1 | 320/320 (100%) |
| Candida glabrataATCC 90030 | ATCC | – | – | Candida glabrata(96.9%) | Candida glabrata | KY102111.1 | 326/326 (100%) | Candida glabrata | KY106473.1 | 347/347 (100%) |
| Candida parapsilosis ATCC 22019 | ATCC | – | – | Candida parapsilosis(95.2%) | Candida parapsilosis | KY102315.1 | 320/320 (100%) | Candida parapsilosis | KY106671.1 | 331/331 (100%) |
| Candida tropicalisNRRL Y-12968 | NRRL | – | – | Candida tropicalis(92.3%) | Candida tropicalis | KY102481.1 | 348/348 (100%) | Candida tropicalis | KY106848.1 | 382/382 (100%) |
BAL: bronchoalveolar lavage; PF: peritoneal fluid; ETA: endotracheal aspirate. *: Strains with discordant results between API ID32C identification and genotypic identification.
Reference strains were from: ATCC, American Type Culture Collection; NRRL, Northern Regional Research Laboratory Collection.
The isolates were identified by conventional methods: colony morphology, germ tube test, and microscopic morphology on corn meal agar with Tween® 80; carbohydrate assimilation tests were performed using the commercial API ID32C® system according to the manufacturer's instructions (bioMérieux, Marcy-l’Etoile, France). The isolates were stored at −80°C in vials containing 0.9% NaCl until use.
DNA isolationGenomic DNA was extracted from the isolates using a modified phenol–chloroform–isoamyl alcohol method. Briefly, after thawing, each isolate was cultured on Sabouraud dextrose agar (Oxoid, Basingstoke, UK) and incubated for 48h at 35°C. One loop of yeast cells was suspended in 100μl of lysis solution [10mM Tris–HCl (pH 8.0), 10mM EDTA (pH 8.0), 50mM NaCl, and 2% SDS], 10μl of proteinase K solution (100mg/ml) were added, and the mixture was incubated for 2h at 65°C. Proteinase K was inactivated by heating to 95°C for 5min. The supernatant was collected, and the suspension was treated with phenol–chloroform–isoamyl alcohol (25:24:1, v/v/v). Subsequently, the samples were extracted with chloroform–isoamyl alcohol (24:1, v/v), and DNA was precipitated with a 2.5X volume of ethanol. DNA was eluted in 30μl of Tris–EDTA (TE) buffer; the DNA concentrations and the A260/280 ratio were determined using a NanoDrop ND-1000 spectrophotometer (ThermoScientific, Wilmington, DE, USA). Samples were stored at −20°C until use.
PCR amplificationTo amplify the ITS region, fungus-specific universal primers ITS1 and ITS4 were used. The D1/D2 region of the 26S rRNA gene was amplified using NL1 and NL4 primers (Table 2). Amplification reactions were performed in a total volume of 25μl containing 2.5μl of 10X PCR buffer, 2.5μl of MgCl2 (25mM), 0.5μl of dNTP mixture (10mM), 0.5μl of each primer (10pmol/μl), 0.3μl of Taq DNA polymerase (5U/μl) (Fermentas, Vilnius, Lithuania), and 75ng of DNA. PCR was carried out in a GeneAmp PCR system 2700 (Applied Biosystems, Inc., Foster City, CA, USA) using the following conditions: initial denaturation at 95°C for 10min, followed by 35 cycles of denaturation at 95°C for 15s, annealing at 55°C (ITS1 and ITS4) or 60°C (NL1-NL4) for 1min, elongation at 72°C for 30s, and a final extension at 72°C for 5min.
Primers used in PCR and DNA sequencing.
| Subunit or spacer region | Name of the primer | Oligonucleotide sequence (5′-3′) |
|---|---|---|
| ITS1-5.8S-ITS2 rDNA | ITS1 (forward) | 5′-TCC GTA GGT GAA CCT GCG G-3′ |
| ITS4 (reverse) | 5′-TCC TCC GGT TAT TGA TAT GC-3′ | |
| D1/D2 region of LSU rDNA | NL1 (forward) | 5′-GCA TAT CAA TAA GCG GAG GAA AAG-3′ |
| NL4 (reverse) | 5′-GGT CCG TGT TTC AAG ACG G-3′ | |
The PCR products were enzymatically purified using ExoSap-IT (USB Corporation, Cleveland, OH, USA). Sequencing reactions were performed using a modified protocol of the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Briefly, 3.2pmol of forward or reverse primers, 4μl of BigDye™ Terminator Ready Reaction mix, 4μl of sequencing buffer, and 4μl of purified amplified DNA (25ng/μl) were combined to a final volume of 20μl. Thermal cycling parameters were as follows: 1min at 96°C, followed by 25 cycles of 10s at 96°C, 5s at 50°C, and 4min at 60°C. Sequencing reaction products were purified using Sephadex G-50 (Sigma–Aldrich, St. Louis, MO, USA). The purified product was analyzed on an automated ABI PRISM 310 Genetic Analyzer (Applied Biosystems). The sequences and electropherograms obtained were analyzed using Sequencing Analysis 5.3.1 software (Applied Biosystems). The sequences were compared to the reference data available from the GenBank database using the BLASTN tool of the National Center for Biotechnology Information database.
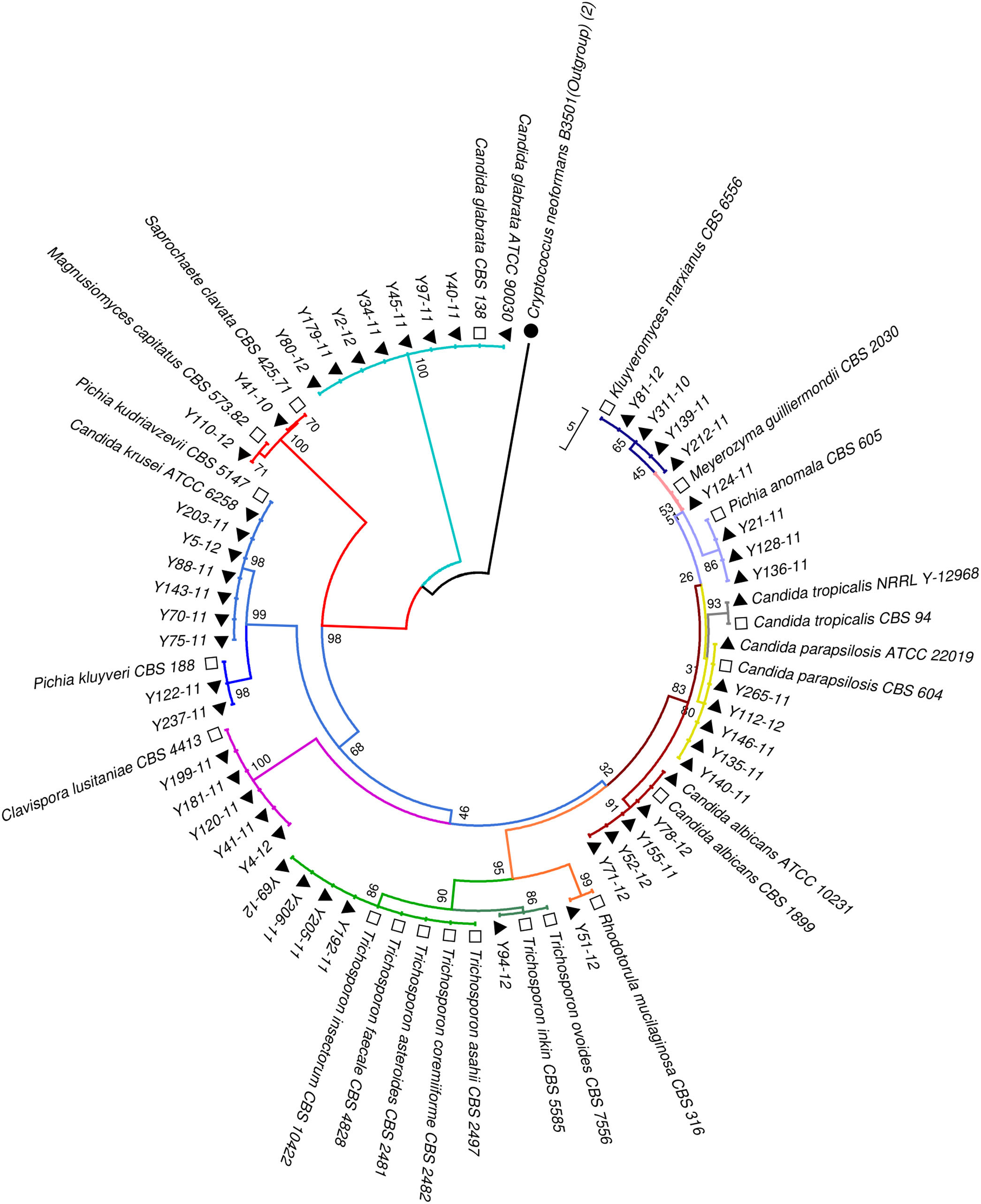
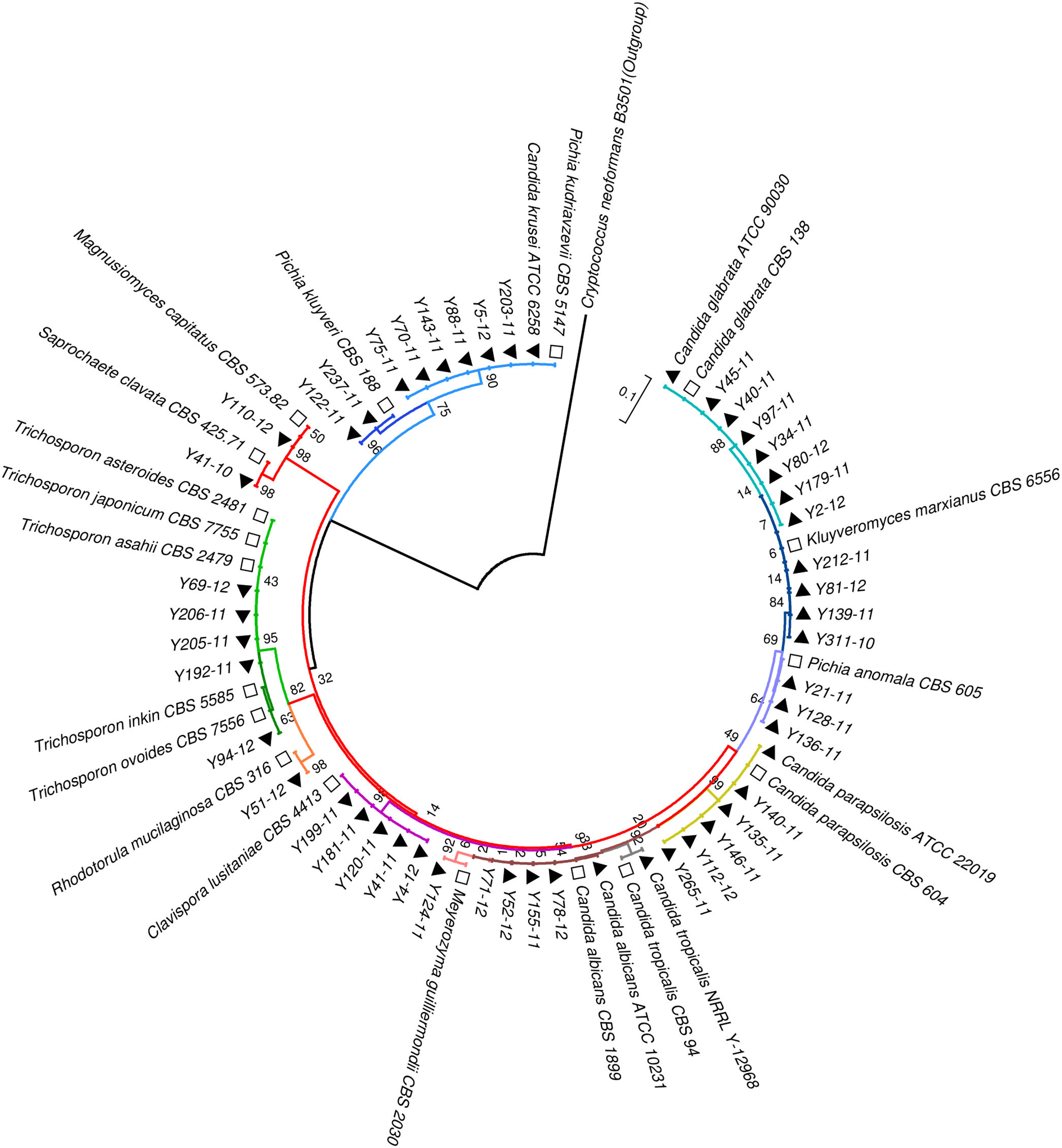
Phylogenetic analysisAll the sequences of the ITS and D1/D2 regions were aligned using the CLUSTALW program, and a phylogenetic tree was constructed using MEGA version 7.0 software. The neighbor-joining algorithm and Tamura-Nei model were implemented. All the gaps were excluded from the analysis, and branch support was ascertained using 1000 bootstrap replicates.13 (Figs. 1 and 2).
 was used as an out-group during phylogenetic analysis.")
Neighbor-joining tree based on the ITS region sequences showing the phylogenetic relationship between the yeast isolates. Bootstrap values were computed over 1000 replicates. Black triangles mark the strains sequenced in this study. White squares represent CBS reference strains. Cryptococcus neoformans B3501 (black circles) was used as an out-group during phylogenetic analysis.
 was used as an out-group during phylogenetic analysis.")
Neighbor-joining tree based on the D1/D2 region sequences showing the phylogenetic relationship between the yeast isolates. Bootstrap values were computed over 1000 replicates. Black triangles mark the strains sequenced in this study. White squares represent CBS reference strains. Cryptococcus neoformans B3501 (black circles) was used as an out-group during phylogenetic analysis.
The statistical software SPPS 19 (IBM SPSS Statistics for Windows, Version 20.0. Armonk, NY: IBM Corp. Released 2011) was used to analyze the data. A concordance analysis was performed to assess the compatibility between the different methods used in the fungal identification. The overall accuracy and weighted kappa (wK) values were calculated. Weighted kappa measure was used to assess agreement between two methods as follows: κ<0, poor agreement; κ=0–0.20, slight agreement; κ=0.21–0.40, fair agreement; κ=0.41–0.60, moderate agreement; κ=0.61–0.80, substantial agreement; and κ=0.81–1.00, almost perfect agreement.5
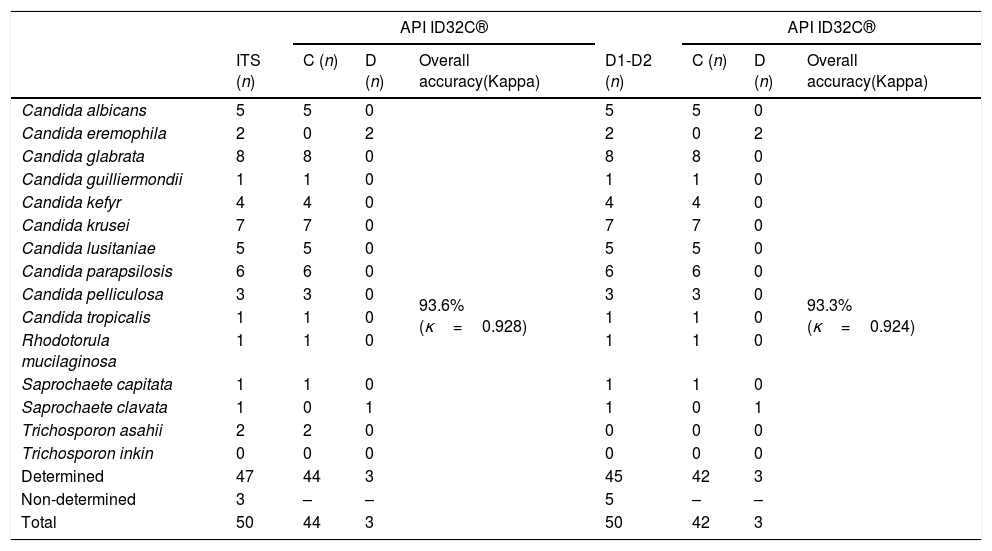
ResultsAmong the 50 strains studied, 5 were reference strains and 45 were clinical yeast isolates. The forty-five clinical yeast isolates were obtained from 28 (62.2%) male and 17 female (37.8%) patients. The patients’ age ranged from 1 to 86 years, with a mean of 57.06 years. The clinical findings in the patients, as well as the results of the phenotypic and molecular identification for the two genetic targets, are shown in Table 1. The 50 isolates were identified using the phenotypic methods as follows: five were identified as C. albicans, seven as C. krusei, five as Candidalusitaniae, six as C. parapsilosis, nine as C. glabrata, two as C. tropicalis, one as Candida guilliermondii, four as Candida kefyr, three as Candida pelliculosa, two as Saprochaete capitata, one as Rhodotorula mucilaginosa, four as Trichosporon asahii, and one as Trichosporon inkin. Table 3 shows the concordance values obtained for the phenotypic and genotypic identifications. The concordance analysis to compare the API ID32C® method against the sequencing analysis of ITS and D1/D2 gave the following wK results: vs. ITS, κ=0.928; vs. D1/D2, κ=0.924 (Table 2). By comparing these methods, all reference strains and isolates phenotypically identified as C. albicans, C. krusei, C. lusitaniae, C. parapsilosis, C. guilliermondii, C. kefyr, C. pelliculosa, and R. mucilaginosa showed 100% concordance with their molecular identifications. However, there was a discrepancy between the phenotypic and the molecular identification for three isolates: two Candida eremophila isolates (Y122-11, Y237-11) were misidentified as Candida glabrata and Candida tropicalis, respectively, and one Saprochaete clavata isolate (Y41-10) was misidentified as S. capitata (Tables 1 and 3).
Comparison of concordances and discrepancies of the API ID32C® against the ITS and D1/D2 sequencing.
| API ID32C® | API ID32C® | |||||||
|---|---|---|---|---|---|---|---|---|
| ITS (n) | C (n) | D (n) | Overall accuracy(Kappa) | D1-D2 (n) | C (n) | D (n) | Overall accuracy(Kappa) | |
| Candida albicans | 5 | 5 | 0 | 93.6%(κ=0.928) | 5 | 5 | 0 | 93.3%(κ=0.924) |
| Candida eremophila | 2 | 0 | 2 | 2 | 0 | 2 | ||
| Candida glabrata | 8 | 8 | 0 | 8 | 8 | 0 | ||
| Candida guilliermondii | 1 | 1 | 0 | 1 | 1 | 0 | ||
| Candida kefyr | 4 | 4 | 0 | 4 | 4 | 0 | ||
| Candida krusei | 7 | 7 | 0 | 7 | 7 | 0 | ||
| Candida lusitaniae | 5 | 5 | 0 | 5 | 5 | 0 | ||
| Candida parapsilosis | 6 | 6 | 0 | 6 | 6 | 0 | ||
| Candida pelliculosa | 3 | 3 | 0 | 3 | 3 | 0 | ||
| Candida tropicalis | 1 | 1 | 0 | 1 | 1 | 0 | ||
| Rhodotorula mucilaginosa | 1 | 1 | 0 | 1 | 1 | 0 | ||
| Saprochaete capitata | 1 | 1 | 0 | 1 | 1 | 0 | ||
| Saprochaete clavata | 1 | 0 | 1 | 1 | 0 | 1 | ||
| Trichosporon asahii | 2 | 2 | 0 | 0 | 0 | 0 | ||
| Trichosporon inkin | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Determined | 47 | 44 | 3 | 45 | 42 | 3 | ||
| Non-determined | 3 | – | – | 5 | – | – | ||
| Total | 50 | 44 | 3 | 50 | 42 | 3 | ||
C: Concordances; D: Discrepancies.
In this study, four out of the five Trichosporon isolates (two from immunosuppressed patients) were identified as T. asahii and one as T. inkin using ID32C. Two of the four isolates identified as T. asahii by ID32C (Y205-11, Y69-12) were 100% identical to T. asahii CBS 2479 according to sequence analysis of the ITS region (379/379 and 428/428, respectively). The ITS sequences of one Trichosporon isolate (Y192-11) in this study was 100% identical (379/379) to that of T. asahii CBS 2479, with a 1-bp difference distinguishing it from the ITS sequence of Trichosporon asteroides CBS 6183 (378/379, 99%) and Trichosporon coremiiforme CBS 2482 (378/379, 99%). The other Trichosporon isolate (Y206-11) had an ITS sequence 100% identical (438/438) to that of T. asahii CBS 2479, with 1-bp difference distinguishing it from the ITS sequence of Trichosporon faecale CBS 4828 (437/438, 99%) and Trichosporon insectorum CBS10421 (437/438, 99%). Four isolates identified as T. asahii by phenotypic methods were 100% identical to T. asahii CBS 2479, Trichosporon japonicum CBS 8641, and T. asteroides CBS 7624 using analysis of the D1/D2 region (Table 1). The isolate identified as T. inkin by ID32C (Y94-12) was found to be 99% identical to T. inkin CBS 7613 and Trichosporon ovoides CBS 5580 by analysis of the ITS region (441/444 and 439/444, respectively), whereas analysis of the D1/D2 region revealed 100% sequence identity with both T. inkin CBS 7613 (385/385) and T. ovoides CBS 5580 (381/385) (Table 1).
DiscussionIn the present study, we employed rRNA gene sequencing to identify medically important yeast species, investigated the performance of the two commonly used amplification targets (the ITS and D1/D2 regions), and compared these methods with conventional methods. Many commercial yeast identification kits are currently available; the API ID32C® or VITEK systems are commonly used to identify yeasts. The VITEK system has the advantage of speed over the API ID32C® system (incubation in 1–15h vs. 48–72h). The API ID32C® kit has a relatively large database for 69 species; however, additional tests are required to identify 11 of the 69 species.14,21,22 Studies comparing DNA sequence analysis and commercial yeast identification methods have reported that the latter techniques may be misleading, being unable to identify less common pathogens and failing to distinguish closely related species.4,15,22,25
Aiming to evaluate the effectiveness of ITS region sequence analysis using 113 yeast isolates, Ciardo et al.4 compared molecular identification and phenotypic description using the API ID32C® system for yeast strains that cannot be identified on the rice agar or CHROMagar Candida medium. Using sequence analysis, 98% of the strains were identified correctly at the species level. Using the API ID32C® system, 87% of all strains were identified correctly at the species or genus level, 7% were undefined, and 6% were misidentified, most of which were reported to be Candida rugosa or Candida utilis. Using the germ tube test, the AUXACOLOR2 system and API20C, Linton et al.15 identified 2880 (95%) of 3033 clinical isolates belonging to 50 different yeast species sent to the UK mycology reference laboratory between 2004 and 2006. The remaining 153 isolates (5%), encompassing 47 species, could not be identified by the available conventional methods and were subjected to molecular identification; using sequence analysis of the D1/D2 region were finally identified. The authors concluded that this method was a reliable technique complementary to conventional methods when identifying rare clinical yeast species.
Sanguinetti et al.25 evaluated 750 clinical isolates with the VITEK 2 and RapID Yeast Plus identification systems, and used sequence analysis of the ITS region as a reference method. The VITEK 2 and RapID systems accurately identified 737 (98.2%) and 716 (95.5%) isolates, respectively. The VITEK 2 system misidentified nine isolates (C. albicans, C. glabrata, C. krusei, C. lipolytica, C. parapsilosis, and Sacharomyces cerevisiae), and didn’t identify four isolates (C. albicans). The authors concluded that sequence analysis of the ITS region is useful to resolve contradictory results from phenotype-based species identification.
The findings in our study are in agreement with those of previous studies. The API ID32C® showed high concordance with ITS and D1/D2 sequencing. The analysis of concordance between these technologies showed an almost perfect agreement (vs. ITS, κ=0.928; vs. D1/D2, κ=0.924), but there were discrepancies in the identification of 3 of the 50 analyzed isolates. Two isolates (Y122-11 and Y237-11) identified as C. glabrata and C. tropicalis, respectively, using API ID32C® were identified as C. eremophila using sequence analysis of the ITS and D1/D2 regions. C. eremophila is an uncommon yeast not included in the API ID32C® database. In two cases—one of malignant neoplasm of the nasopharynx and the other of interstitial lung disease—the yeasts from the sputum culture were identified as S. capitata using API ID32C®, and arthrospores and blastospores were microscopically observed when grown on corn meal agar with Tween® 80. According to sequence analysis of the ITS and D1/D2 regions, one of the isolates (Y110-12) was S. capitata, and the one that belonged to the patient with nasopharynx malignant neoplasm (Y41-10) was identified as S. clavata, which is an ascomycete closely related genetically to S. capitata. The latter causes invasive infections in patients with hematological malignancies, whereas S. clavata is a very rare, but emerging causative agent of invasive human infections.3,7 In this study, S. clavata was isolated from a patient for the first time in Turkey. Similar to C. eremophila, S. clavata is not found in the API ID32C® database.
Most Trichosporon species are found commonly in the natural environment (soil, water, and plants); however, they may also be part of the normal flora of human skin, respiratory tract, and gastrointestinal and genitourinary systems.10 Today, the Trichosporon genus is composed of 50 species and five different clades (Brassicae, Cutaneum, Gracile, Ovoides, and Porosum). Sixteen of the 50 species are clinically significant, and eight of them (T. asahii, T. asteroides, Trichosporon cutaneum, T. inkin, Trichosporon mucoides, T. ovoides, T. japonicum, and Trichosporon loubierii) are considered infectious and allergic agents. The most common cause of invasive Trichosporon infections in the world is T. asahii, and infections caused by Trichosporon have been associated with high mortality rates, despite antifungal therapy.10,24 The correct characterization of Trichosporon species is of therapeutic importance since different species have different antifungal susceptibilities. T. asahii is more resistant to amphotericin B, and T. faecale and T. coremiiforme exhibit higher minimum inhibitory concentration values for amphotericin B than other species.6,24,30 Databases of the existing VITEK 2 and API ID32C® systems are limited to T. asahii, T. inkin, and T. mucoides, and therefore they are not sufficiently reliable to identify Trichosporon species, which may result in the misidentification of genetically different species.5,30
Sequence variation in the ITS and D1/D2 regions has been used in previous researches for species assignment.28,29 However, as the Trichosporon species are phylogenetically closely related to each other, several researchers have reported that sequence analyses of the ITS and D1/D2 regions were unable to clearly discriminate between all the species in this genus.6,14,24,30 Sugita et al.28 first introduced sequence analysis of the IGS (intergenic spacer) region as a useful method to identify closely related Trichosporon species, and reported more variation in the IGS1 region than in the ITS region in the comparative analysis of nucleotide sequences. It has been shown in the Trichosporon genus that when the nucleotide sequence difference between the IGS regions of two strains is <50%, their difference in the ITS region is only 2%. The authors concluded that sequence analysis of the IGS region is a powerful method to distinguish between phylogenetically closely related species. Guo et al.8 compared conventional phenotypic methods and DNA sequence analysis of the ITS, D1/D2, and IGS regions to identify 45 clinical and three reference Trichosporon isolates. They reported that, all 48 isolates were identified successfully by sequencing their ITS and IGS regions. D1/D2 region correctly identified 47 of 48 (97.8%) strains, with the exception of one strain, which was identified as T. mucoides but as Trichosporon dermatis by ITS and IGS sequencing. The commercial API20C AUX and VITEK2 Compact YST systems correctly identified 91.9% and 73% of isolates of the species included in the databases, and misidentified 63.6% and 36.4% of isolates of species not included in the databases, respectively. The authors also stated that one of the two T. japonicum isolates was identified as T. asahii by commercial identification systems, and that this misidentification had also occurred after the first reported isolation of T. japonicum.
Similarly to previous studies, we were unable to distinguish among the species of Trichosporon using sequence analysis of the ITS and D1/D2 regions (Table 1). On the other hand, the sequence variation of the IGS region may be used to successfully discriminate between phylogenetically related Trichosporon species. However, the lack of sequencing of the IGS region for Trichosporon species and the low number of isolates are among the limitations of our study.
The factors that lead to choose certain tests in the clinical laboratory are cost-effectiveness and turnaround time. The costs of the two methods used in this work were, approximately, 9 € for API ID32C® kit, and 40 € for the bi-directional sequencing analysis. Using sequence analysis requires a turnaround of 12h, whereas the use of API ID32C® requires 24–48h.
Although, ITS and D1/D2 sequencing is unquestionably the gold standard to identify yeasts, the API ID32C® presented high concordance with the ITS and D1/D2 sequencing. In conclusion, since it is cost-effective, API ID32C® can be used to identify pathogenic yeasts that are more commonly seen in clinical laboratories as the first-line evaluation tool. We recommend the use of sequence analysis in cases where phenotypic methods are insufficient to identify the species (e.g. rare species), where rapid diagnosis has a crucial impact on the patient (e.g., isolation of fungus from sterile body fluids), and during epidemiological research that requires species-level identification.
Conflict of interestsThe authors declare no conflict of interests.
This research was supported in part by the State Planning Organization Foundation (Turkey) (Grant No: 2008K120640).

