





Current methods for the laboratory diagnosis of histoplasmosis are problematic in terms of their sensitivity, specificity and runtime.
ObjectivesThus, in this study, we sought to select and optimize methods for the detection of Histoplasma capsulatum var. capsulatum by polymerase chain reaction (PCR).
MethodsThree DNA extraction methods and three PCR methods were evaluated. We optimised the concentration of the components of this PCR reaction and determined its sensitivity and specificity using blood samples to which H. capsulatum had been added.
ResultsThe DNA extraction method that yielded the highest-quality DNA used silica membranes (DNeasy Blood & Tissue Kit, Qiagen, Hilden, Germany), and the amplification method with the best detection capacity used a target gene encoding a 100-kDa protein. Our optimisation of the PCR conditions indicated that the reaction works over a significant range of component concentrations; in addition, it was able to detect H. capsulatum better than traditional culture techniques, with a detection limit of only 10 pg of DNA.
ConclusionsIn our experimental conditions, the PCR method selected in this work (instead of nested-PCR) is a tool sensitive enough for the diagnosis of histoplasmosis.
Los métodos actuales para el diagnóstico de laboratorio de la histoplasmosis son problemáticos si consideramos los valores de sensibilidad, especificidad y tiempo de ejecución.
ObjetivosEn este estudio hemos tratado de seleccionar y optimizar los métodos para la detección de Histoplasma capsulatum var. capsulatum mediante la reacción en cadena de la polimerasa (PCR).
MétodosSe evaluaron tres métodos de extracción de ADN y tres métodos de PCR. Se optimizó la concentración de los componentes de esta reacción de PCR y se determinó su co-positividad (sensibilidad) y co-negatividad (especificidad), utilizando muestras de sangre a las que se había añadido H. capsulatum.
ResultadosEl método de extracción que dio el ADN de más alta calidad utiliza membranas de sílice (DNeasy Blood & Tissue Kit, Qiagen, Hilden, Germany), y el método de PCR con mayor capacidad de detección es el que incluye un gen diana que codifica una proteína de 100 kDa. Nuestra optimización de las condiciones de PCR indicaron que la reacción trabaja en un rango significativo de las concentraciones de componentes y, además, fue capaz de detectar H. capsulatum mejor que las técnicas de cultivo tradicional, con un límite de detección de solo 10 pg de ADN.
ConclusionesEn nuestras condiciones experimentales, el método PCR seleccionado en este trabajo (en lugar de PCR anidada) es una herramienta lo suficientemente sensible para el diagnóstico de la histoplasmosis.
Histoplasmosis is a systemic mycosis caused by the fungus Histoplasma capsulatum var. capsulatum. The prevalence of this disease has increased over the years6; however, while the majority of infections are self-limiting, this fungus can lead to an acute, chronic or disseminated infection in immunocompromised patients.14,17 Disseminated histoplasmosis is the most serious form of the disease and occurs primarily in immunosuppressed patients, especially those with HIV/AIDS.10,23,33
Histoplasmosis is diagnosed based on direct microscopic examination, culturing and the detection of antigens and antibodies.21,27,32,33 However, direct microscopic examination, with or without specific histopathological staining, is only positive in approximately 50% of cases.6,20,21 In addition, H. capsulatum grows slowly in culture, and its identification takes three to six weeks.8,16,29 Laboratory manipulation is dangerous and should be performed in a biosafety level III facilities.2,21,29 The sensitivity of culturing tests varies depending on the type of the sample analysed. With bone marrow, the sensitivity is 70% to 90%, whereas with respiratory specimens it is 50-90%. Although methods that detect antibodies are much faster than culture, their sensitivity depends on the immunity of the patient, the stage of the disease and its clinical form.6,17,25 Finally, although detecting the H. capsulatum antigen in biological samples is a sensitive and specific method (95% sensitivity in urine samples), it is very expensive and not currently available in most laboratories.31
Given these issues, new methods have been developed to improve the laboratory diagnosis of histoplasmosis. Among these methods, PCR assays have been evaluated as possible diagnostic tools.2,4,11,21,24 However, these methods are not yet used in mycology laboratories due to the lack of standardised protocols for extracting and detecting Histoplasma DNA.
Recently, three authors have proposed methods for detecting H. capsulatum using PCR. These methods are based on detecting genes encoding specific proteins (a 100-kDa protein,2 the M antigen4 and the H antigen15). Two of these methods are nested PCR reactions that have a high detection capacity and high sensitivity but require a more steps than regular PCR and give a higher rate of false positives.22
Because studies comparing DNA extraction methods and PCR detection of Histoplasma capsulatum var. capsulatum are scarce, we have screened and optimised methods for detecting this organism by PCR.
Materials and methodsFungal isolatesWe used three Histoplasma capsulatum isolates (FMT3417, FMT3342 and FMT3378) from the fungal collection of the Fundação de Medicina Tropical do Amazonas. These strains were sub-cultured and turned into their yeast and filamentous phases as described by Imano et al.19
Selecting a DNA extraction methodWe compared three DNA extraction methods: a) extractions based on the use of silica membranes (DNeasy Blood & Tissue Kit, Qiagen, Hilden, Germany), b) extractions based on the use of salt precipitation (MasterPure DNA Purification Kit for Blood Version II, Epicentre, Madison, WI, USA), and c) phenol-chloroform DNA extractions based on polarity fractions.30 We followed the manufacturers’ instructions when using commercial DNA extraction kits. We modified the phenol-chloroform extractions30 to use glass beads (0.45mm) for cell disruption. The best method for extraction was determined using the data of DNA concentration and purity. We determined the concentration of genomic DNA by spectrophotometry (GeneQuant pro RNA/DNA Calculator, GE Healthcare, Piscataway, NJ, USA) at 260nm (one unit of absorbance corresponded to 50μg/ml). The ratio of the absorbances at 260nm and 280nm was used to determine the purity of the DNA.
Selecting a PCR methodThe DNA obtained with the best extraction method (previous item) was submitted to PCR. We evaluated three PCR-based methods for the detection of Histoplasma capsulatum (Table 1). Samples with different concentrations of H. capsulatum DNA (25, 0.5, 1×10−2, 2×10−4, 4×10−6 and 8×10−8 ng) were used for amplification in order to determine the detection limit of each protocol. We analysed the samples by electrophoresis on a 1.5% agarose gel as described by Bialek et al.2
PCR protocols investigated.
| Author/Target Region | Reaction conditions |
| Bialek et al., 2002/Protein 100 KDa | Primers: HcIII (5-GAG ATC TAG TCG CGG CCA GGT TCA-3) andHcIV (5-AGG AGA GAA CTG TAT CGG TGG CTT G-3)Product size: 210 bp[DNTPs]: 100μM[Taq polymerase]: 1.5 U[MgCl2]: 2.5 mM[Primers]: 1μM[Amplification temperatures]: 94°C for 5 min; 35 cycles of 94°C for 30 s, 65°C for 30 s and 72°C for 1 min, and a final extension of 72°C for 5 min |
| Bracca et al., 2003/ Antigen H | Primers: Hc2 (5-GCGGGGTTGGCTCTGCTCT-3) andHc3 (5-TTGGAAACCCCGGGCTTG-3)Product size: 439 bp[DNTPs]: 200μM[Taq polymerase]: 1 U[MgCl2]: 2 mM[Primers]: 0.4μM[Amplification temperatures]: 96°C for 6 min; 35 cycles of 94°C for 1 min, 59°C for 1min, and 72°C for 1 min, and a final extension of 72°C for 10 min |
| Guedes et al., 2003/Antigen M | Primers: Msp1F (ACAAGA GACGACGGTAGCTTCACG) andMsp1R (GCGTTGGGGATCAAGCGATGAGCC)Product size: 111 bp[DNTPs]: 200μM[Taq polymerase]: 2.5 U[MgCl2]: 1.5 mM[Primers]: 0.8μM[Amplification temperatures]: 95°C for 5 min, 35 cycles of 95°C for 1 min, 70°C for 1 min and 72°C for 1 min, and a final extension of 72°C for 5 min |
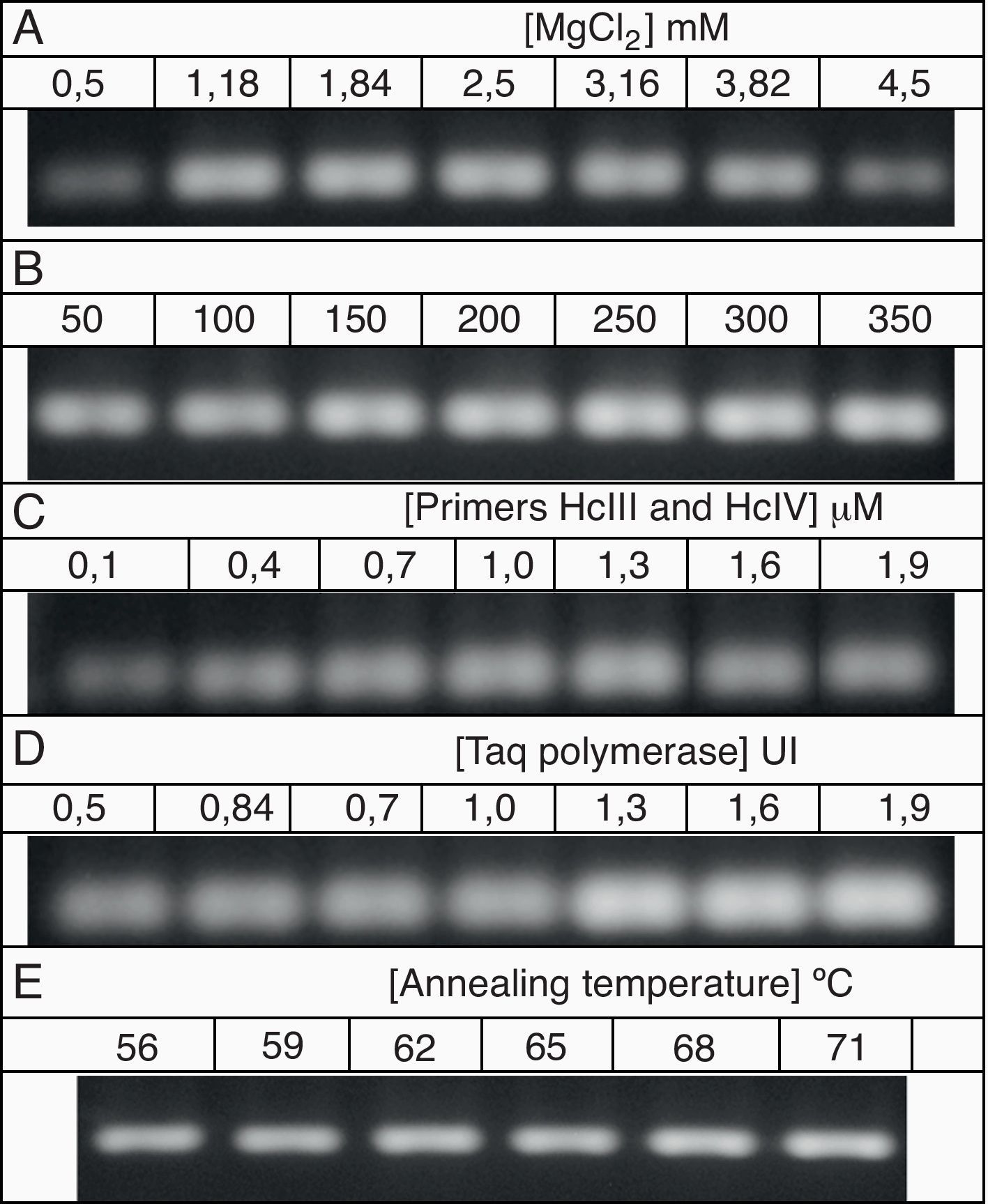
The PCR protocol selected in the previous item was submitted to optimization using a univariate method. The components and their concentrations were as follows: 1) MgCl2 (0.5, 1.18, 1.84, 2.5, 3.16, 3.82 and 4.5mM), 2) dNTPs (50, 100, 150, 200, 250, 300 and 350μM), 3) Primers HcIII and HcIV (0.1, 0.4, 0.7, 1.0, 1.6 and 1.9μM), 4) Taq DNA polymerase (0.5, 0.7, 0.84, 1.0, 1.3, 1.6 and 1.9 U per reaction), and 5) annealing temperature (56, 59, 62, 65, 68 and 71°C). Each reaction had a final volume of 25μl, including 50 ng of DNA template.
Determining the sensitivity of the PCRSensitivity of the optimized PCR (previous item) was determined as described by Maubon et al.,25 using fifteen peripheral blood samples from a healthy volunteer. We added a known concentration of H. capsulatum yeast cells to the samples (7×106, 7×105, 7×104, 7×103 and 7×102 cells/mL) from three different isolates (FMT3417, FMT3342 and FMT3378). All of the samples were used to detect H. capsulatum both by the previously described PCR screening methods and by blood culture.29 Sample volumes of 200μL were used for the detection assays.
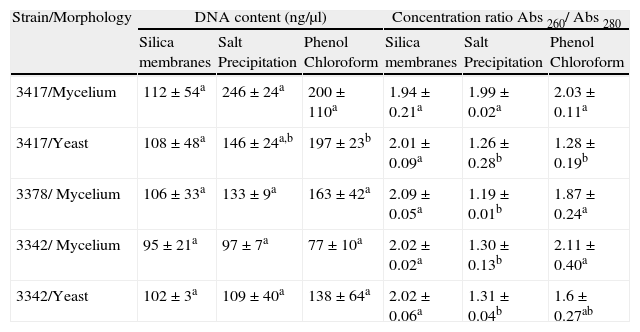
ResultsWe evaluated three methods for extracting DNA from H. capsulatum. Each method was fundamentally different in its means of nucleic acid isolation (use of silica membranes, precipitation of components with salt or separation based on polarity). Table 2 shows the concentrations and levels of purity of the DNA extracted using each method.
Concentration and purity ratio of H. capsulatum DNA obtained by different extraction methods.
| Strain/Morphology | DNA content (ng/μl) | Concentration ratio Abs 260/ Abs 280 | ||||
| Silica membranes | Salt Precipitation | Phenol Chloroform | Silica membranes | Salt Precipitation | Phenol Chloroform | |
| 3417/Mycelium | 112±54a | 246±24a | 200±110a | 1.94±0.21a | 1.99±0.02a | 2.03±0.11a |
| 3417/Yeast | 108±48a | 146±24a,b | 197±23b | 2.01±0.09a | 1.26±0.28b | 1.28±0.19b |
| 3378/ Mycelium | 106±33a | 133±9a | 163±42a | 2.09±0.05a | 1.19±0.01b | 1.87±0.24a |
| 3342/ Mycelium | 95±21a | 97±7a | 77±10a | 2.02±0.02a | 1.30±0.13b | 2.11±0.40a |
| 3342/Yeast | 102±3a | 109±40a | 138±64a | 2.02±0.06a | 1.31±0.04b | 1.6±0.27ab |
The media, reported with the same letters, have no significant difference between them at a level of 95%. Testing performed by ANOVA and Fisher test.
The three methods resulted in similar concentrations of DNA. The silica membrane-based extraction method resulted in the purest DNA (260/280nm) for fungal samples in the yeast phase. For the isolates FMTAM 3342 and FMTAM 3378, the salt-precipitation method of extraction resulted in lower levels of purity than the other methods.
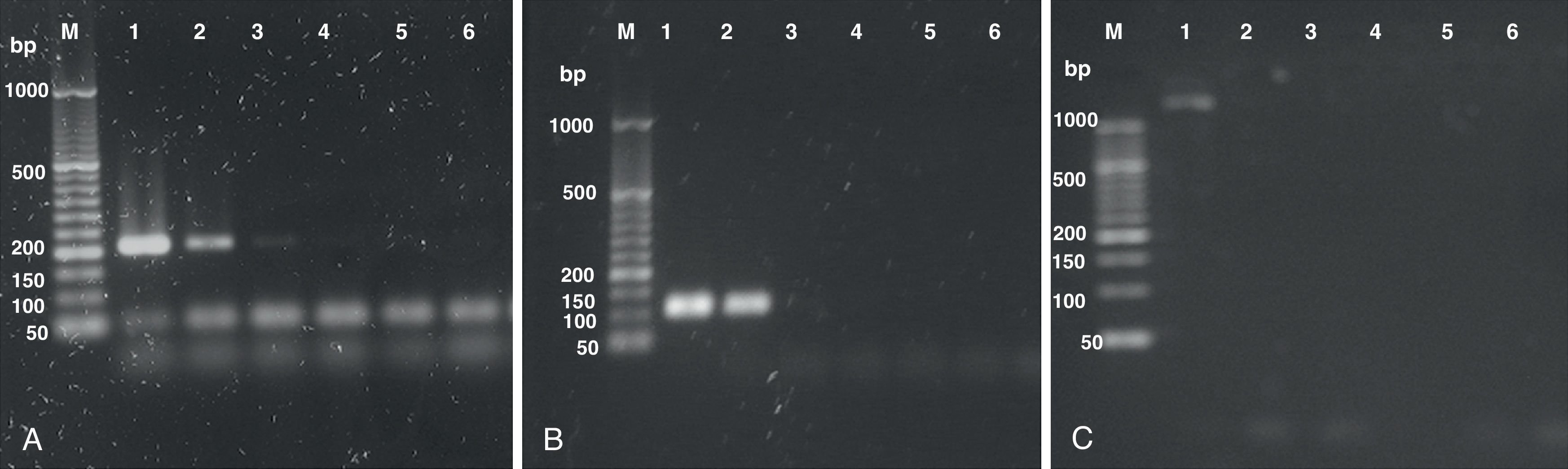
Samples containing different concentrations (25 ng, 0.5 ng, 1×10−2, 2×10−4, 4×10−6 and 8×10−8 ng) of H. capsulatum DNA were used to evaluate the ability of the PCR methods described by Bialek et al.,2 Guedes et al.15 and Bracca et al.4 to detect H. capsulatum. These methods involve targeting DNA sequences codifying for a 100-kDa protein, the M antigen and the H antigen, respectively, for amplification. The results from our evaluation of these PCR methods are shown in Figure 1.
, Guedes15 (B) and Bracca4 (C). 50bp marker (M), 25 ng (1), 0.5 ng (2), 1×10−2 (3), 2×10−4 (4), 4×10−6 (5) and 8×10−8ng (6) of DNA.")
The lowest concentrations of DNA that yielded PCR products using the methods described by Bialek et al.2 and Guedes et al.15 were 0.01 and 0.5 ng, respectively. The method described by Bracca et al.4 did not yield the 439 bp PCR product described by the authors; however, it presented a large (> 1000bp) non-specific band. This PCR protocol was repeated several times without success and then discarded.
We selected the silica membrane DNA extraction method and the modified PCR protocol of Bialek et al.2 for using in the remainder of this work. The concentrations of the PCR reagents were optimised using univariate analysis to improve the method's detection capacity.
All of the optimisation reactions resulted in PCR products that could be adequately visualised after agarose gel electrophoresis (Fig. 2). Primer annealing temperatures of 56°C and 59°C resulted in a non-specific band of approximately 100bp (data not shown). The conditions considered optimal that were used for subsequent analyses were as follows: 2.5mM MgCl2, 200μM dNTPs, 1.0μM of each primer, 1 U Taq polymerase and an annealing temperature of 71°C.

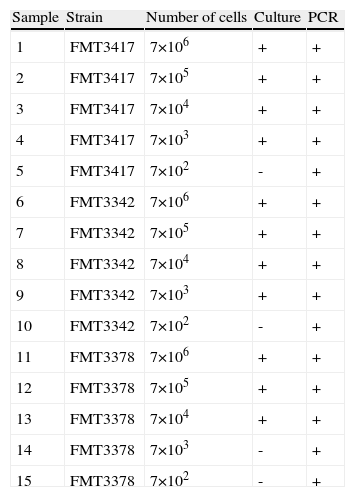
We used fifteen blood samples to evaluate the sensitivity and specificity of the PCR and blood culture methods. We made five cell suspensions in human blood (7×106, 7×105, 7×104, 7×103 and 7×102 cells/ml) from each of three different H. capsulatum isolates (FMT3417, FMT3378 and FMT3342).
All of the blood samples containing H. capsulatum cells were positively identified by PCR, regardless of the concentration of the fungal cells (Table 3). We also found that 11 out of the 15 contaminated samples tested positive by blood culture. The three samples containing 7×102 cells/mL (samples 10, 20 and 30) and one of the samples containing 7×103 cells/mL (sample 28) did not grow in culture after 40 days of incubation. These results were confirmed by repetition of the assays.
Results of PCR and culture for the detection of H. capsulatum in blood samples.
| Sample | Strain | Number of cells | Culture | PCR |
| 1 | FMT3417 | 7×106 | + | + |
| 2 | FMT3417 | 7×105 | + | + |
| 3 | FMT3417 | 7×104 | + | + |
| 4 | FMT3417 | 7×103 | + | + |
| 5 | FMT3417 | 7×102 | - | + |
| 6 | FMT3342 | 7×106 | + | + |
| 7 | FMT3342 | 7×105 | + | + |
| 8 | FMT3342 | 7×104 | + | + |
| 9 | FMT3342 | 7×103 | + | + |
| 10 | FMT3342 | 7×102 | - | + |
| 11 | FMT3378 | 7×106 | + | + |
| 12 | FMT3378 | 7×105 | + | + |
| 13 | FMT3378 | 7×104 | + | + |
| 14 | FMT3378 | 7×103 | - | + |
| 15 | FMT3378 | 7×102 | - | + |
It is possible to detect H. capsulatum by PCR when the DNA from the pathogen, present in a biological sample, is obtained at a sufficient concentration and purity.8 The three DNA extraction methods evaluated in this study yielded similar concentrations of extracted DNA (Table 2). Einsele et al.,11 Bernal et al.1 and Reed et al.28 achieved similar results in fungi, bacteria and protozoans, respectively. This finding justifies, even today, the existence of various methods for the extraction of genomic DNA.
Because the DNA extraction methods produced similar final concentrations of DNA, we also evaluated the number of steps, the DNA purity and the time needed to extract the DNA. The DNA obtained using the Qiagen kit was the purest one (260nm/280nm), and this was also the method that required the least amount of time. This result corroborates the findings of studies performed by Einsele et al.,11 Reed et al.28 and Zetzsche et al.,34 in which purer DNA (260nm/280nm) was obtained with silica membrane-based methods. The silica membrane specifically binds to nucleic acids during the extraction procedure, allowing the nucleic acids to be separated from other cellular components.34
The salt precipitation extraction method resulted in the least pure DNA. This method involves the precipitation of proteins in acetate salts and the recovery of DNA by precipitation in isopropyl alcohol.13 However, the acetate salts are not specific for proteins, and, under the conditions studied, this method was not adequate for our purposes.
The sensitivity, specificity and detection limit of PCR depend on the primers used. Appropriate sequences that are directed at a target region of the pathogen's DNA must be used.5,12,22 The genetic regions designated as the “100-kDa protein”, H antigen and M antigen have been described by Bialek et al.,2 Bracca et al.4 and Guedes et al.,15 respectively, as being appropriate regions for detecting H. capsulatum. Bialek et al.2 and Bracca et al.4 used nested PCR as an analytical tool. However, in our study we decided to modify these techniques to evaluate whether a single PCR reaction could be used for diagnostic analysis, as nested PCR assays have contamination problems that are difficult to prevent in the daily routine of a laboratory.
Under our experimental conditions, the best method for detecting H. capsulatum was that of Bialek et al.,2 which we adjusted to require a single PCR rather than nested reactions. It should be noted that of the genetic regions we evaluated, the 100-kDa protein region was the most well-studied, as other studies have successfully used this region to detect H. capsulatum in biological samples.9,25,31 There is limited information regarding the copy numbers of the genes we evaluated, and our focus on optimizing methods did not allow us to address this matter in any depth.
In our study, the lowest concentration of DNA that we were able to amplify using the method described by Guedes et al.15 was 500 pg. This result differs from that described by the authors, who obtained PCR products with as little as 1 pg of DNA. This difference may be due to the microorganisms used,7,19 the quality of our sample DNA and/or general laboratory conditions. In the case of the methods described by Bracca et al.,4 despite numerous repetitions and primer substitutions, we could not produce the PCR products described by the authors. Further studies are needed to understand the reasons for this disparity.
Although some authors have been able to reproduce the nested PCR methods described by Bialek9,25,31 using biological samples, our study was the first to optimise the concentrations of the PCR components. Optimisation studies, which present the usual concentrations of each component and demonstrate the problems that can arise with higher or lower concentrations of each reaction component, are important and widely discussed in the literature.3,18,22,26,32
Specifically, in the experiments performed in this study, it was difficult to determine which concentrations of PCR components worked best because there were several PCR products with similar intensities after gel electrophoresis. This result is promising because it demonstrates that the PCR reactions were able to generate sufficient products under all of the conditions tested in this experiment. Furthermore, these results demonstrate that the values previously established by Bialek et al.,2 which have been reproduced in other studies, are sufficient for detecting H. capsulatum.9,25,31This is the first study to demonstrate that PCR with the primers HcIII and HcIV has a limit of detection higher than that of traditional culture techniques and also to support the utilization of a “simple PCR” instead of nested PCR for detection of H. capsulatum. (Table 3). Using this reaction, we were able to detect DNA concentrations as low as 10 pg. Thus, this method can be used to detect the pathogen in biological samples, positive liquid cultures (blood cultures, myeloma cultures) and in “young” cultures (that cannot be phenotypically characterised) and even to support a previous diagnosis.
Authors’ declarationWe take the opportunity to declare that: a) the contents of the article are original and they were not published previously; b) there is no conflict of interests, related to financial aspects; c) all the authors have read and approved this manuscript, and d) this work was previously submitted and approved by the Research Ethics Committee of the Tropical Medicine Foundation of Amazonas.
The authors would like to thank Dr. Júlia Salem-INPA for her structural support, involvement and corrections as well as the Instituto Nacional de Pesquisas da Amazônia (INPA) (National Research Institute of Amazônia) for allowing us to use their facilities for optimising the PCR detection assays in the Mycobacteriology Laboratory-CPCS. We are grateful to FAPEAM for their financial support through the grant PPSUS 2006.

