



Las malformaciones craneofaciales son algunas de las patologías más prevalentes en la edad pediátrica. Podemos distinguir dos grandes grupos: las producidas por un cierre precoz de las suturas del esqueleto craneofacial, las craneosinostosis y faciocraneosinostosis; y las que actualmente se pueden considerar neurocrestopatías, como los síndromes de primer y segundo arcos branquiales y las fisuras orofaciales como la fisura labiopalatina.
Se describen las principales características de los síndromes más frecuentes y los tratamientos, desde los más empleados a los más innovadores, como las técnicas endoscópicas y las de distracción ósea.
Craniofacial abnormalities are some of the most prevalent malformations in children. We can distinguish two groups: those caused by an early closure of the sutures of the craniofacial skeleton, as craniosynostoses and faciocranioynostosis; and those that are considered neural crest anomalies, like the first and second brachial arch syndromes, and cleft lift and palate.
The authors discuss the major characteristics of the most frequent syndromes, as well as treatment modalities, including the most popular and the most recent ones, like endoscopic and bone distraction techniques.
Las malformaciones craneofaciales son algunas de las patologías más prevalentes en la edad pediátrica. Algunas de ellas, como las craneales, pueden poner en peligro la vida del niño o dejar secuelas irrecuperables como el déficit intelectual. Por otro lado, las malformaciones faciales no suelen suponer un riesgo vital; sin embargo, marcan a los niños y a sus familias de por vida. La mayoría de ellos necesitarán múltiples y complejas operaciones para intentar que su apariencia facial llegue a ser lo más adecuada posible.
La amplia variedad de anomalías craneofaciales muchas veces las hace inclasificables, Gorlin et al (1, 2) concluyen que esta limitación corresponde a la falta de comprensión embriológica y a las causas que las provocan. En 1981 se reúne el Comité de Nomenclatura y Clasificación de las Anomalías Craneofaciales derivada de la Asociación Americana de Fisura Labiopalatina. El comité propuso una clasificación simple, dividida en cinco categorías:
- I-
Fisuras Faciales/ Encefaloceles/ Disostosis
- II-
Atrofia/ Hipoplasia
- III-
Neoplasias
- IV-
Craneosinostosis
- V-
Inclasificables
Esta simple clasificación nos permite tener un resumen y orden de las causas y manejo de las anomalías craneofaciales más frecuentes (3).
Embriología craneofacialLa embriología del desarrollo craneofacial fue descrita por Sperber (4). Las células de la cresta neural de la zona craneal y vagal van a dar lugar al ectomesénquima de la región cráneo-cérvico-facial y arcos branquiales, a partir del cual se diferencian los procesos faciales.
Una de las características más importantes en la formación de la cara la constituyen los desplazamientos y multiplicación celular que dan como resultado la formación de los mamelones o procesos faciales.
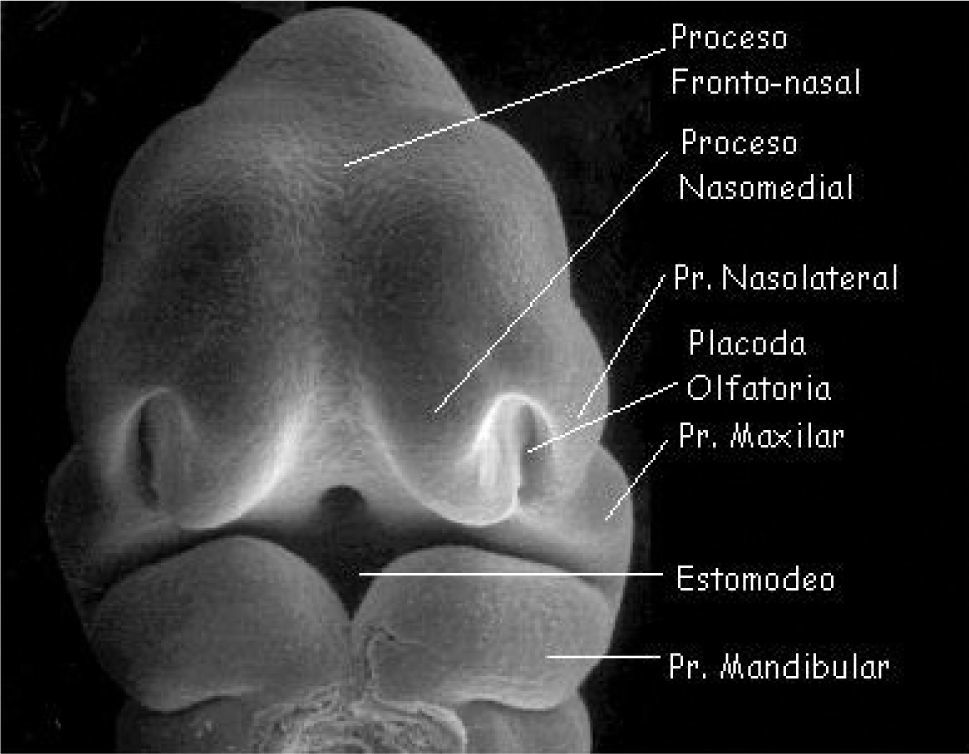
La característica más típica del desarrollo de la cabeza y cuello es la formación de arcos branquiales o faríngeos. Aparecen en la cuarta y quinta semana del desarrollo intrauterino. En un periodo inicial están constituidos por tejido mesenquimático, separados por surcos denominados hendiduras faríngeas. Los arcos branquiales no solo contribuyen a la formación del cuello, sino que desempeñan un papel importante en la formación de la cara, principalmente el primer y segundo arcos (5, 6, 7). La cara se forma entre las semanas cuarta a octava del periodo embrionario gracias al desarrollo de cinco mamelones o procesos faciales: El mamelón cefálico o frontonasal constituye el borde superior del estomodeo o boca primitiva. Los procesos maxilares se advierten lateralmente al estomodeo y, en posición caudal a éste, los procesos mandibulares (ambos procesos derivados del primer arco branquial) (Figura 1).
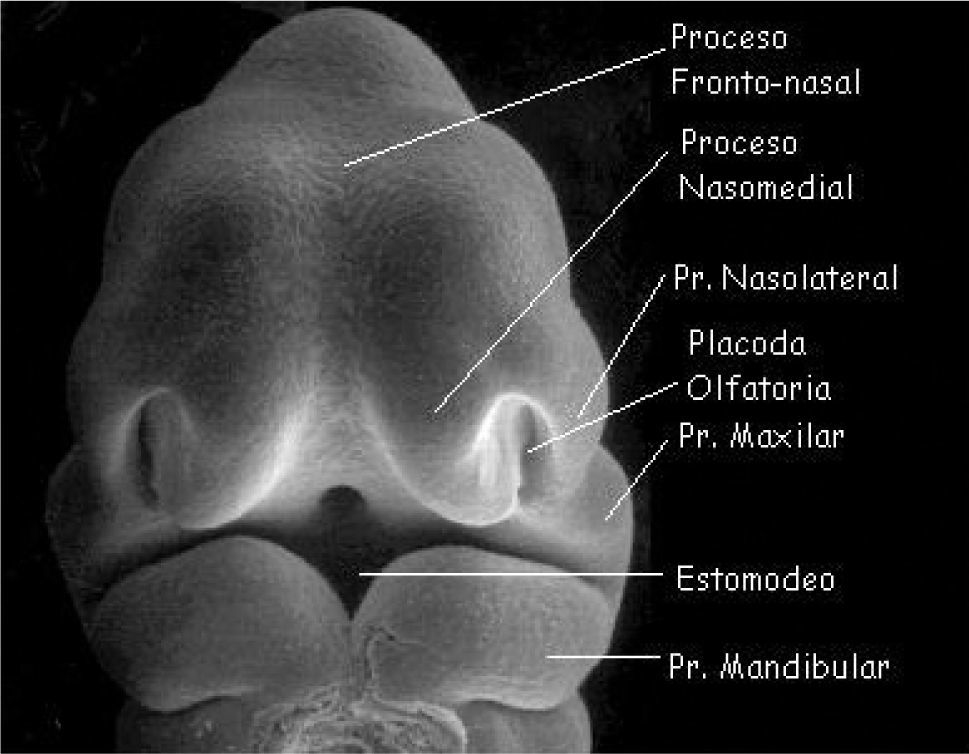
A cada lado de la prominencia frontonasal se observa un engrosamiento local del ectodermo superficial, las placodas nasales u olfatorias. Durante la quinta semana las placodas nasales se invaginan para formar las fositas nasales. En la sexta semana aparecen rebordes de tejido que rodean a cada fosita formando, en el borde externo, los mamelones nasales externos y, del lado interno, los mamelones nasales internos. En el curso de las dos semanas siguientes los procesos maxilares crecen simultáneamente en dirección medial, comprimiendo los procesos nasales hacia la línea media. En una etapa ulterior queda cubierta la hendidura que se encuentra entre el proceso nasal interno y el maxilar, y ambos procesos se fusionan. En consecuencia, el labio superior es formado por los dos procesos nasales internos y los dos procesos maxilares. El labio inferior y la mandíbula se forman a partir de los procesos mandibulares, que se fusionan en la línea media.
La nariz se formará a partir de las cinco prominencias faciales: la prominencia frontonasal da origen al puente de la nariz y la frente; los mamelones nasales externos forman las aletas y los procesos nasales internos fusionados dan lugar a la punta de la nariz.
Las crestas palatinas derivadas de los procesos maxilares se fusionan entre sí la séptima semana, dando lugar al paladar secundario. Hacia delante las crestas se fusionan con el paladar primitivo dejando como línea divisoria entre ambos paladares el agujero incisivo.
El pabellón de la oreja se desarrolla a partir de seis proliferaciones mesenquimatosas en los extremos dorsales del primer y segundo arcos branquiales, y rodeando a la primera hendidura faríngea. Estas prominencias, tres a cada lado del conducto auditivo externo, se fusionan y se convierten poco a poco en la oreja definitiva.
Los ojos comienzan a desarrollarse en forma de un par de vesículas ópticas de cada lado del prosencéfalo al final de la cuarta semana de la vida intrauterina. Las vesículas ópticas, evaginaciones del cerebro, toman contacto con el ectodermo superficial y provocan los cambios necesarios para la formación del cristalino. La córnea se forma a partir de ectodermo superficial y epitelio epidérmico. A través de la fisura coroidea penetra la arteria hialóidea (futura arteria central de la retina) y las fibras nerviosas del ojo.
Causas de anomalias craneofacialesLas malformaciones craneofaciales son las malformaciones congénitas más frecuentes en humanos, pero se sabe muy poco acerca de su etiología. En algunos casos existe una transmisión genética mendeliana, si bien la mayoría son esporádicas. Hay autores que discuten el papel del hipertiroidismo, de algunas metabolopatías, agentes teratogénicos, etc. pero la realidad es que en la mayor parte de los casos la causa es desconocida. El punto de partida y la manera en que progresan son también mal conocidos. En los síndromes asociados con el cierre precoz de suturas se ha demostrado la implicación de ciertos factores de crecimiento o de sus receptores. La base del cráneo y su crecimiento desempeñan un papel muy importante, especialmente en las craneoestenosis con retraso del crecimiento facial.
Últimamente cada vez son más los autores que consideran que muchos de los síndromes con afectación craneofacial tienen algo en común, y es que las malformaciones se producen por alteraciones de las células de la cresta neural y las consideran como neurocrestopatías.
Durante la última década ha existido un gran avance en la identificación de las bases genéticas para la mayoría de los síndromes craneofaciales. Para aquellos casos o condiciones sin un patrón genético identifiable, se han demostrado factores definidos como agentes “teratogénicos”, condicionantes ambientales que se detallan a continuación:
-Radiación. Grandes dosis se asocian a Microcefalia.
-Infección. Neonatos en antecedente de toxoplasma, rubéola o cito-megalovirus tiene una alta incidencia de fisuras faciales.
-Idiosincrasia materna. Niveles altos de fenilketonuria aumenta la incidencia de fisura labiopalatina, hiperinsulinismo se asocia a malformaciones oculoauriculovertebrales y factores como la edad, el peso a otras malformaciones craneofaciales.
-Químicos. Deficiencias vitamínicas se asocia a incrementos en la incidencia de fisura labiopalatinas. Drogas como el tabaco materno y la nitrofurantoina se asocian a craneosinostosis. Alcohol, anticonvulsionantes como la fenitoína y el ácido valproico se asocia a un aumento en la incidencia de fisura labiopalatina (8, 9).
Corresponden las fisuras faciales a las anomalías craneofaciales más frecuentes, siendo la más común aquella que se presenta paralela al filtrum y puede como no comprometer el paladar, también conocida como fisura labio palatina.
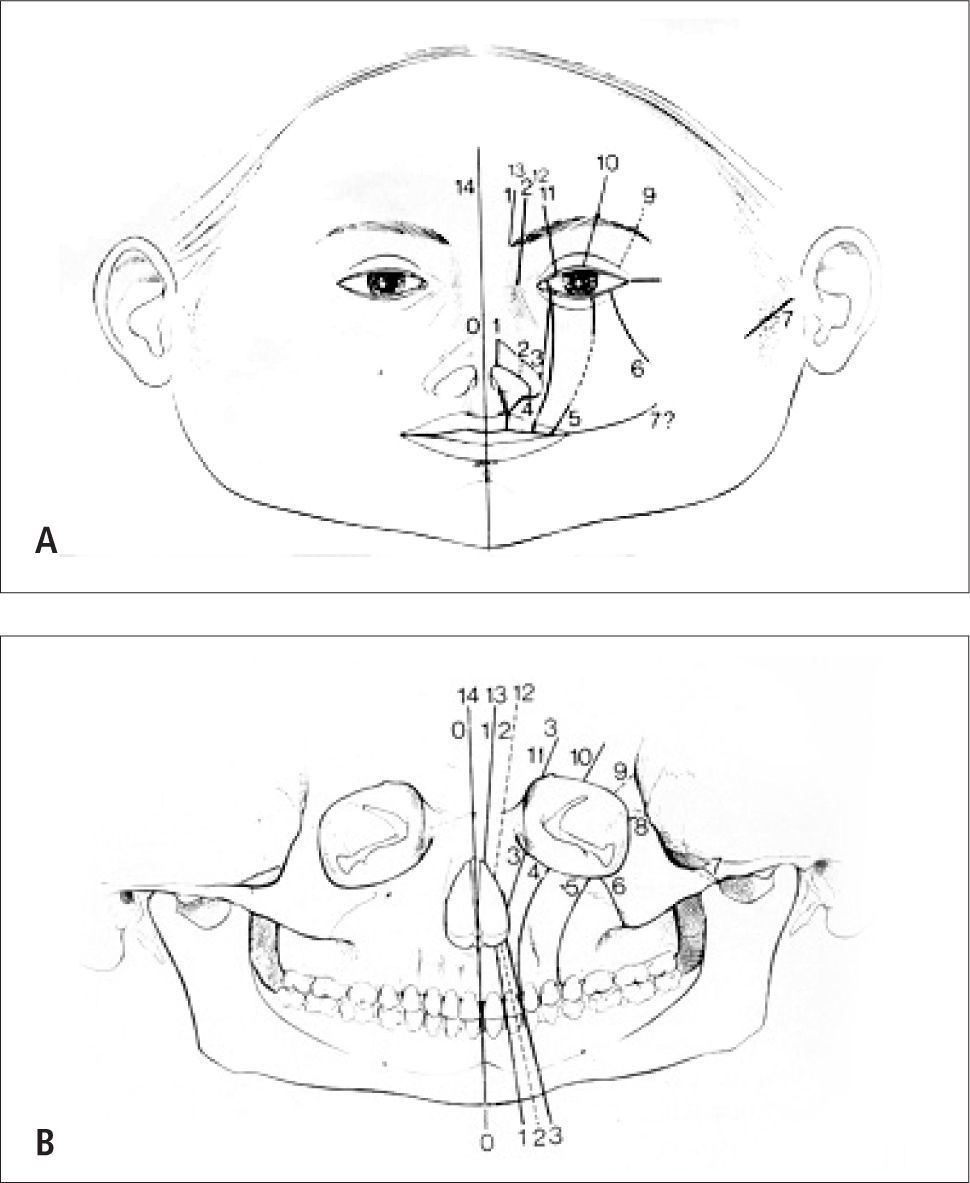
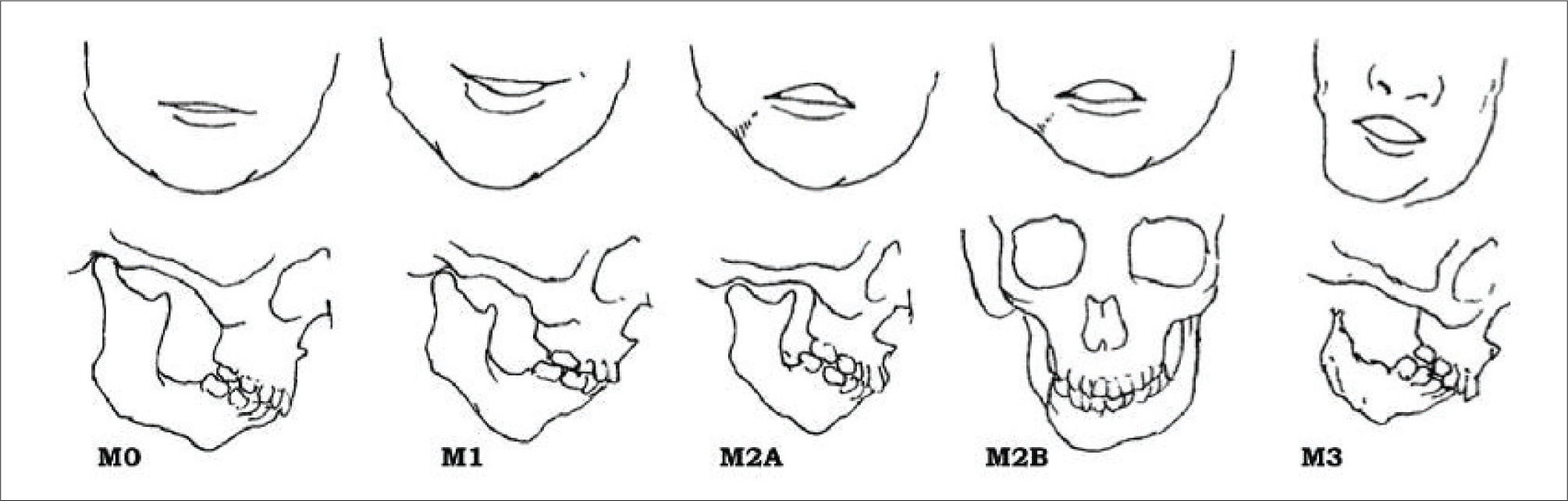
Clasificación AnatómicaEl amplio espectro de anomalías atribuibles a las fisuras faciales convirtió su clasificación en un problema. En 1976, Tessier (10) describió en forma brillante una clasificación con base anatómica, donde le asignó un número correlativo a cada malformación en base a su ubicación en relación a línea media sagital. Esta clasificación fue aceptada en forma internacional y permite una comunicación concisa y efectiva entre los diferentes especialistas (Figura 2A).
")
Para una mejor orientación, la órbita es dividida en dos hemisferios, todo por bajo el párpado inferior corresponden a las fisuras faciales y lo que está sobre el párpado superior a las fisuras craneales. De acuerdo con la clasificación anatómica de Tessier, el compromiso de las partes blandas y su relación con el componente óseo no siempre coinciden, pudiendo incluso coexistir dos o más fisuras.
En resumen las fisuras faciales las podemos numerar de 0 a 14, siendo las fisuras labiopalatinas el 75% de las malformaciones faciales mayores y el 80% de todas las fisuras orofaciales. Estas corresponden según clasificación descrita los números 1–2–3 de Tessier con una incidencia en Chile de 1 cada 700 recién nacidos vivos (Figura 2B).
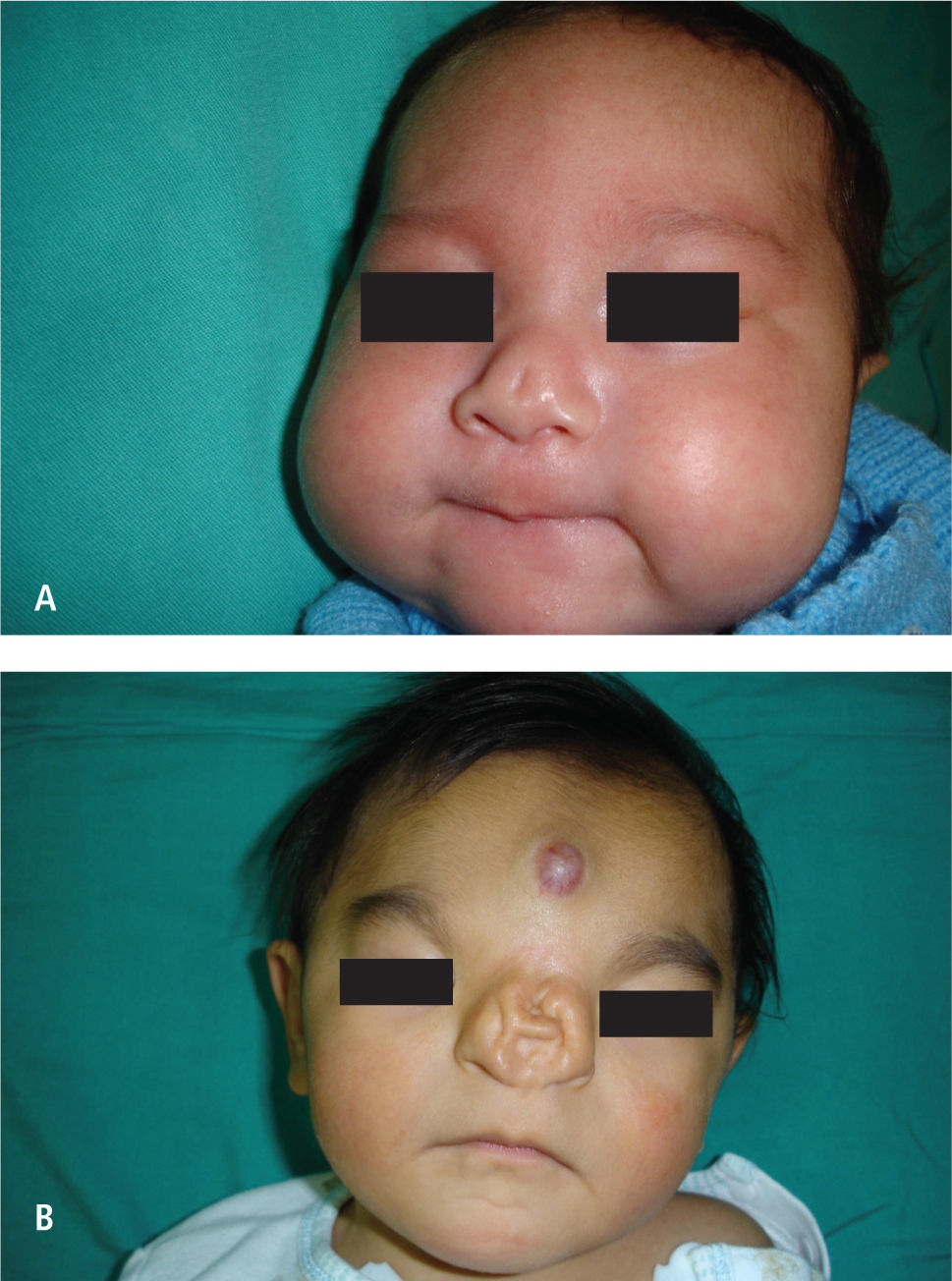
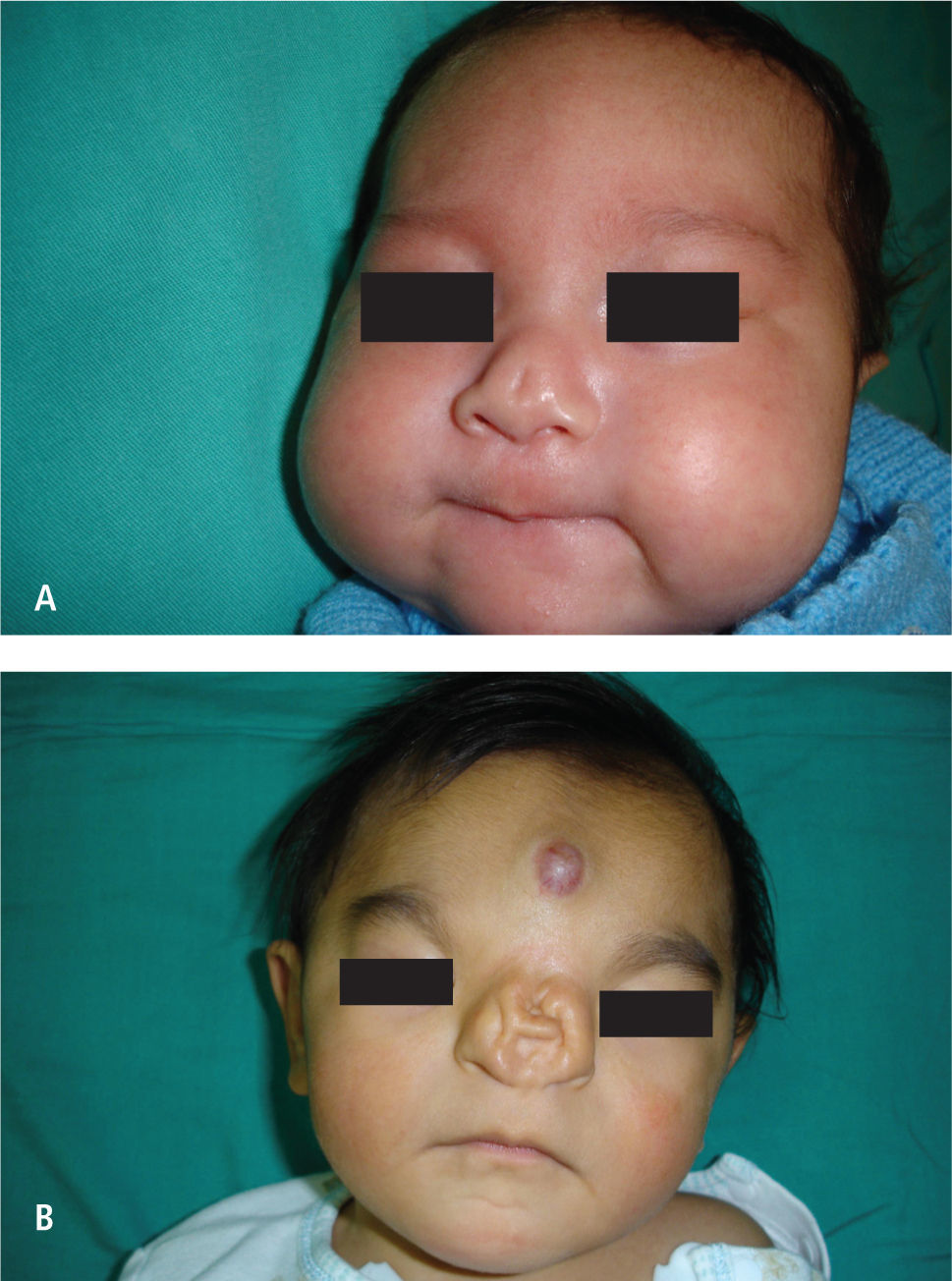
La fisura 0 o fisura media, rara con una incidencia 0.40–0.70%, se ve en menos de cada 100 fisuras. Tiene diferentes grados de expresión y corresponde a la no fusión de los procesos nasales en su origen el problema de este tipo de fisuras es la posibilidad de comprometer según el tipo el desarrollo cerebral, siendo su espectro muy amplio, que va desde un pequeño defecto o “notch” en el bermellón a una fisura media con hipotelorismo con holoproscencefalia (Figura 3).
El tratamiento de las fisuras faciales se enfoca inicialmente en el cierre de las partes blandas (12) con la exéresis del tejido cicatrizal de la fisura, acompañado de un cierre meticuloso y anatómico de los diferentes tejidos involucrados, generalmente si esta se asocia a una insuficiencia esqueletal, su reparación se pospone hasta que el niño sea mayor. En resumen, cada fisura tiene su tratamiento específico, privilegiando siempre los aspectos funcionales primeros, como por ejemplo la reconstrucción palpebral en una exposición del globo ocular, dejando los aspectos cosméticos no menos importantes para etapas posteriores.
IB- EncefaloceleEl encefalocele es una enfermedad rara del desarrollo, del grupo de los defectos en el cierre del tubo neural (tubo longitudinal central del embrión que origina el encéfalo, médula espinal y otros tejidos del sistema nervioso central), lo que normalmente se produce durante la cuarta semana de la gestación; cuando estos defectos en el cierre del tubo neural afectan al cerebro dan lugar a anencefalia y encefalocele y si se localizan en columna vertebral provocan espina bífida.
El encefalocele se caracteriza por herniación o protrusión de parte del encéfalo y de las meninges a través de un defecto craneal; si solamente protruyen las meninges se denomina meningocele craneal, mientras que si protruye el ventrículo se denomina meningohidroen-cefalocele.
El encefalocele es el defecto abierto del tubo neural menos frecuente. Como promedio se presenta entre un caso de cada 2.000 a 6.000 nacidos vivos, pero su incidencia varía considerablemente según los diferentes estudios siendo al parecer más frecuente en Méjico, en países de origen celta y ciertos países del sureste asiático como Indonesia, Malasia y Tailandia, donde llega a alcanzar una frecuencia de uno por cada 5.000 nacidos vivos. Aunque todavía se desconoce su mecanismo de producción, se implican factores genéticos y se estima que aproximadamente el 10% de los defectos del tubo neural son causados por mutaciones genéticas o alteraciones cromosómicas, ya que se ha visto una alta incidencia en hermanos de niños con esta enfermedad.
El contenido típico de la herniación es líquido cefalorraquídeo y tejido neural que se conecta al cerebro a través de un estrecho pedículo; la cubierta del saco herniario puede variar desde una capa bien formada con piel y cabellos a una delgada capa meníngea; por lo que la lesión puede estar totalmente cubierta por piel, o alternar con zonas desprovistas de ésta, que dejan el tejido nervioso al descubierto.
Los encefaloceles se localizan en la región occipital en el 75% de los casos y en menor proporción, alrededor del 15%, pueden localizarse en región parietal frontal y sincipital (sincipucio es la parte anterosuperior de la cabeza) estos últimos se subclasifican por su localización en: nasofrontal, nasoetmoidal y nasoorbital.
Las manifestaciones clínicas dependen de la zona del cerebro herniada, siendo las más frecuentes alteraciones visuales, microcefalia (cabeza anormalmente pequeña), retraso mental y crisis convulsivas; los encefaloceles sincipitales tienen además de las alteraciones visuales, manifestaciones nasales y auditivas.
El encefalocele puede presentarse de forma aislada o asociado a otras anomalías del sistema nervioso central: hidrocefalia, mielomeningocele, ausencia del cuerpo calloso y lisencefalia; a otras malformaciones congénitas: displasia frontonasal, síndrome de bandas amnióticas; también se ha descrito en algunas cromosomopatías trisomía 18 y 13 y delecciones (13q y 16q). Puede formar parte de síndromes polimalformativos como Walker Warburg, síndrome de Meckel, en el que el encefalocele es occipital y menos frecuentemente criptoftalmia de Fraser, síndrome de Knobloch y embriofetopatía por Warfarina.
El diagnóstico diferencial debe tener en cuenta el higroma quístico, en el que no existe ningún defecto óseo, el edema de la calota, teratomas (tumores mixtos complejos, en los que los tejidos múltiples se disponen en órganos diferenciados) y otras anomalías congénitas como anencefalia, hendidura quística braquial, hemangioma y sarcoma mesenquimático. En los casos de encefalocele frontal hay que diferenciarlo del dacriocistocele (quiste del conducto lagrimal) o de un teratoma nasal.
El pronóstico es variable en función por un lado del tamaño la localización y el tipo de tejido cerebral herniado y por otro del número, tipo y severidad de las malformaciones asociadas. Los lactantes con encefalocele tienen más riesgo de presentar una hidrocefalia (acumulación de líquido en el encéfalo) por estenosis (estrechez patológica de un conducto) del acueducto, una malformación de Chiari, o un síndrome de Dandy Walker.
La determinación de niveles de alfafetoproteína materna y la realización de ecografía prenatal, permiten el diagnóstico intraútero que contribuye a un manejo más apropiado del paciente y posibilita el despistaje de otras malformaciones y la planificación del tratamiento. La imagen ecográfica del encefalocele consiste en una masa de tejido asociada siempre a un defecto óseo a través del cual se produce la herniación.
El tratamiento es quirúrgico y debe ser abordado interdisciplinariamente. La mayoría de los encefaloceles deben corregirse, incluso los más grandes ya que puede eliminarse sin provocar incapacidad funcional importante, siendo necesaria la corrección quirúrgica urgente cuando la lesión es abierta, es decir no está cubierta por piel (13,14,15,16,17).
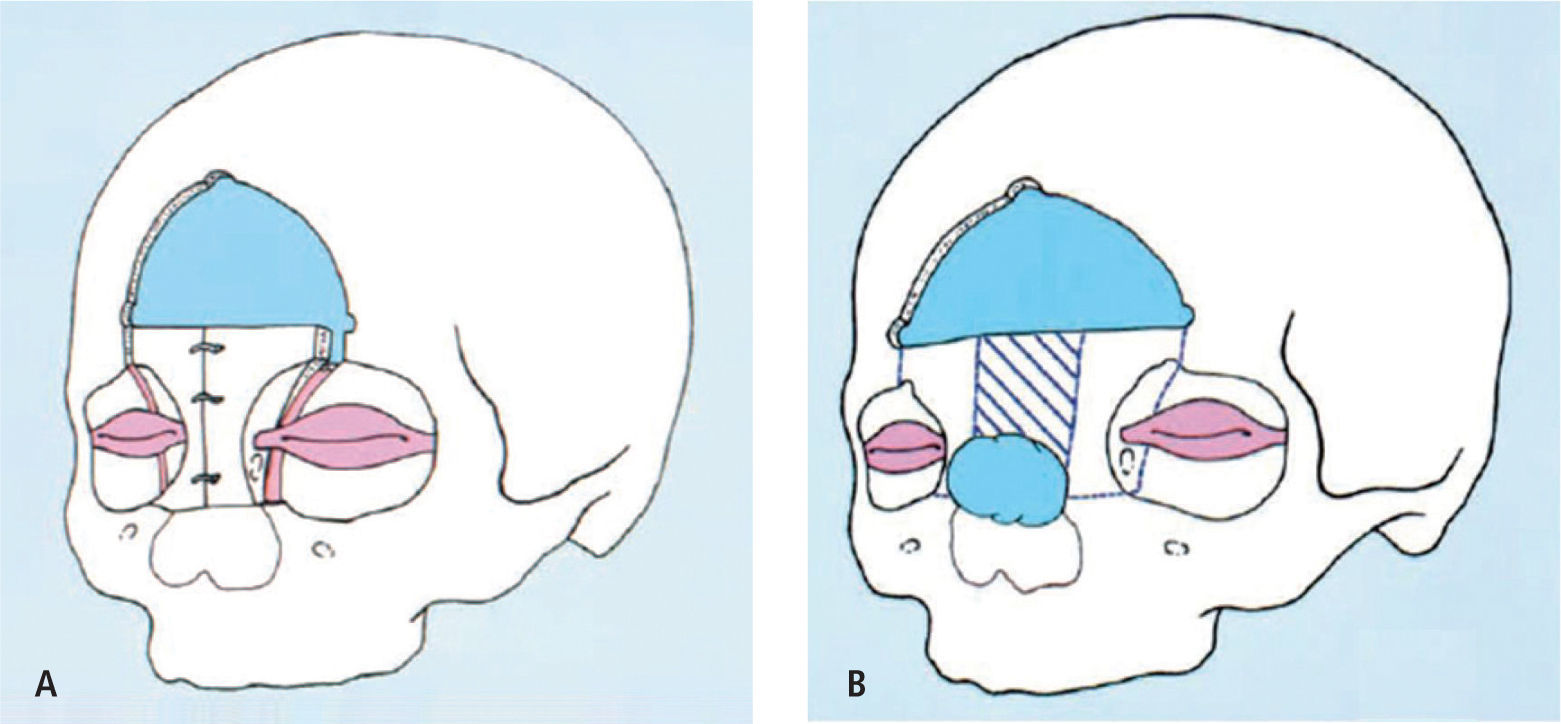
La prevención de los defectos en el tubo neural, se consigue mediante tratamiento con suplementos orales diarios de ácido fólico, suministrados durante el tiempo que transcurre entre la planificación del embarazo y la 12 semana de gestación, por lo que se aconseja comenzar este tratamiento desde el momento en el que se pretenda un embarazo (Figura 4).
IC- Disostosisa) Microsomia hemifacial (MHF)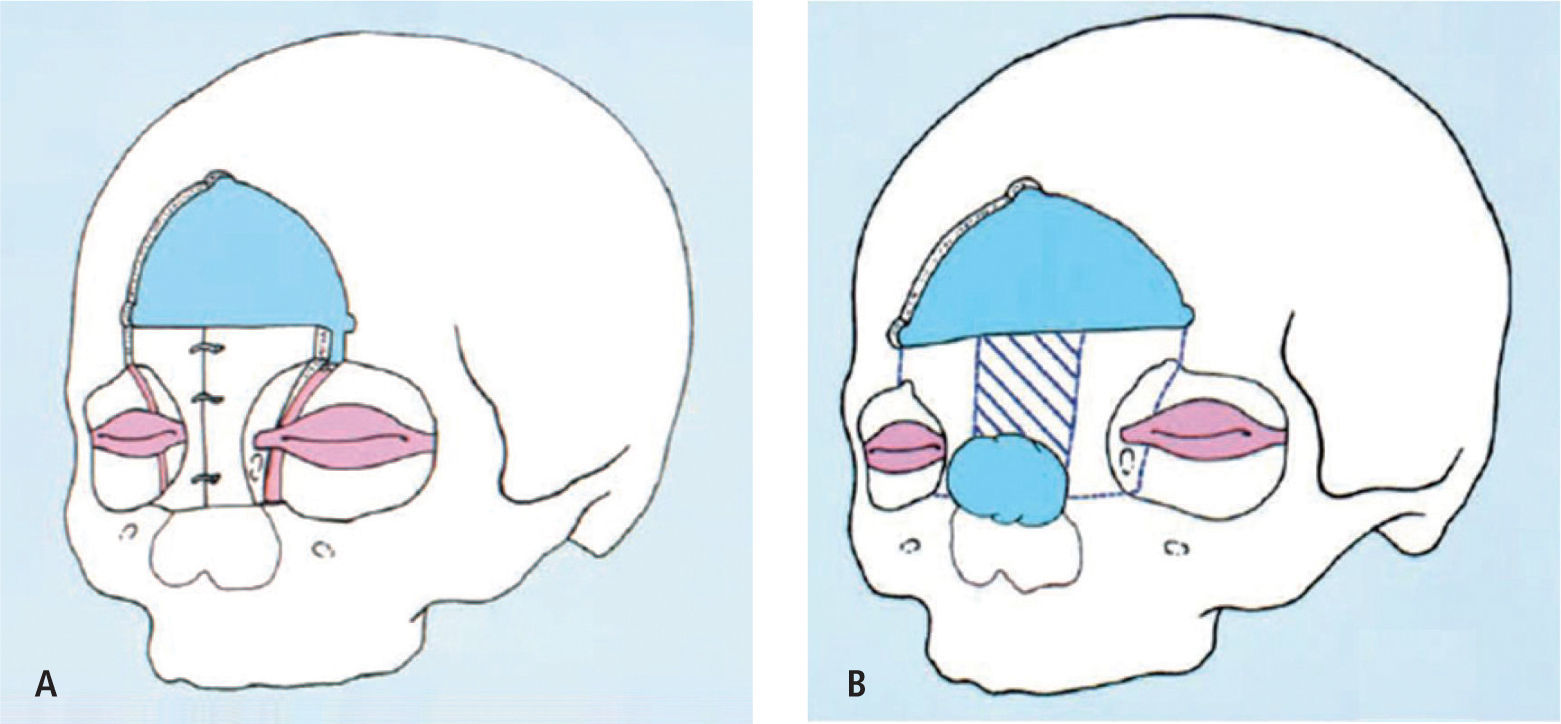
También conocida como síndrome de primer y segundo arco, de correlación directa con la fisura 7 de Tessier. La microsomía hemifacial es un trastorno en el cual el tejido de un lado de la cara no se desarrolla completamente, lo que afecta principalmente las regiones auditiva (del oído), oral (de la boca) y mandibular (de los maxilares). En algunos casos, es posible que ambos lados de la cara se vean afectados e incluso puede haber compromiso de ella y del cráneo. La deformidad en la microsomía hemifacial varía en gran medida según la gravedad del trastorno, que oscila entre leve y grave, y la región de la cara comprometida. Las estructuras que suelen verse comprometidas en diferentes grados son; el oído medio y externo, el maxilar y mandíbula, los dientes, las partes blandas que componen la mejilla y ramos del nervio facial que permiten las expresiones faciales.
La MHF es una entidad con un espectro de anomalías congénitas cuyas lesiones son derivadas del primer y segundo arcos branquiales y que pueden ir asociadas en algunos casos con anomalías extracraneales. Algunos nombres ha tenido, lo que refleja una comprensión incompleta de su patogénesis cuya etiología es heterogénea.
Se han creado algunos sistemas para su diagnóstico. Entre los cuales debemos mencionar la Clasificación de Pruzansky luego modificada por Mulliken, la cual describe la deficiencia mandibular en 3 tipos (Figura 5).
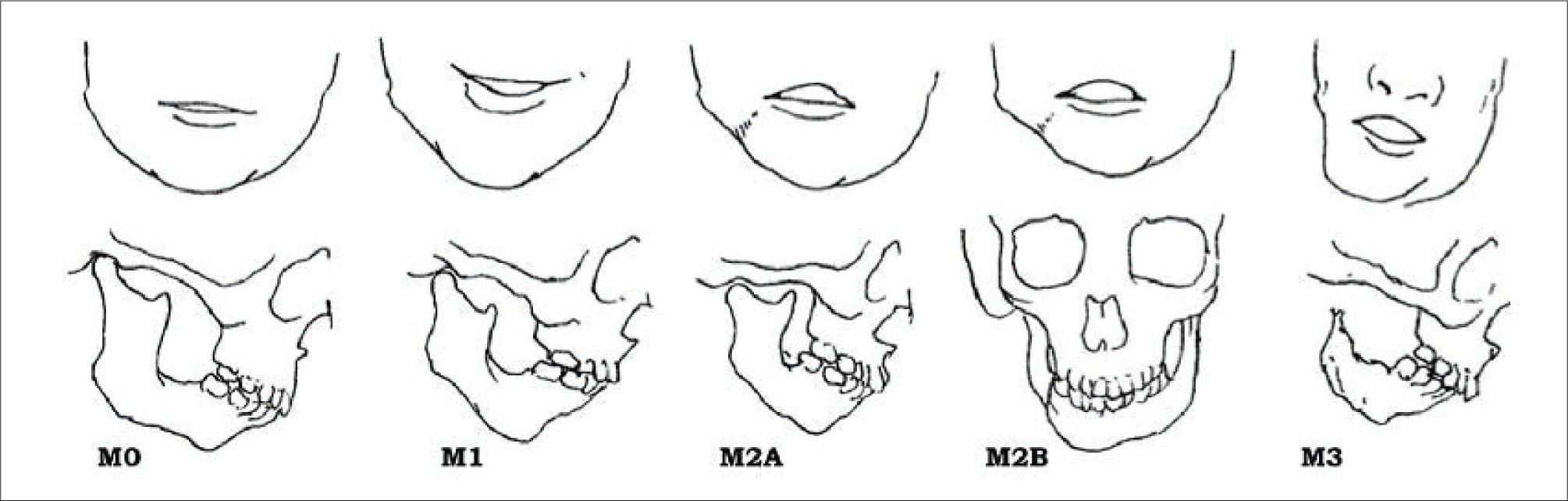
La microsomía hemifacial se diagnostica luego del estudio de los antecedentes médicos y de un examen físico completo realizado por un genetista. No existe un análisis de sangre que permita diagnosticar este trastorno. Debido a que el espectro de gravedad es muy amplio, el diagnóstico debe provenir de un genetista experimentado y capacitado en el diagnóstico de anomalías craneofaciales. También pueden realizarse tomografías computarizadas y radiografías de la cara para obtener un diagnóstico más preciso.
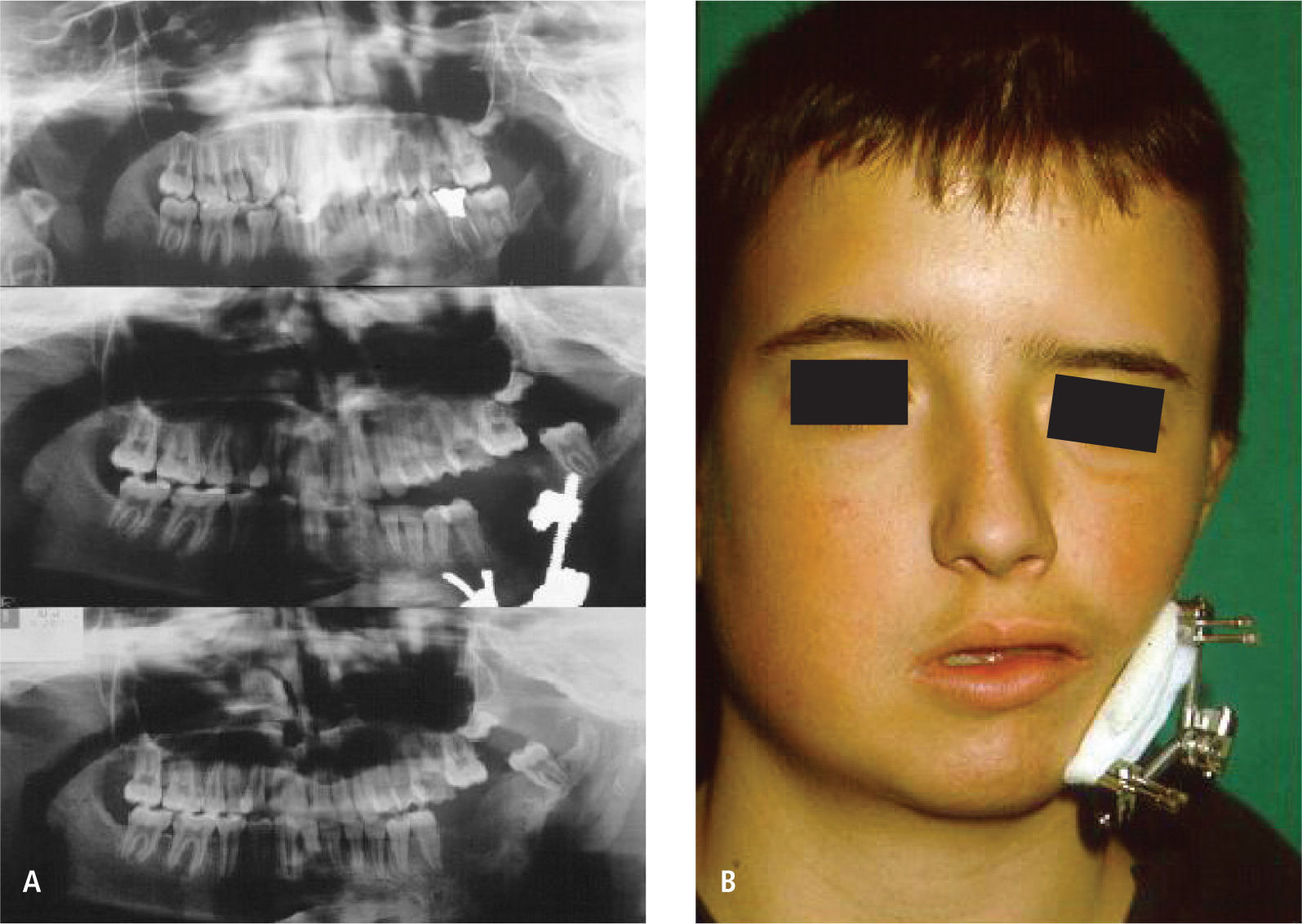
El tratamiento es quirúrgico y multidisciplinario, en función de corregir en forma escalonada aquellas insuficiencias tisulares. En presencia de fisura 7, ésta debe ser corregida durante los primeros meses de vida y según el grado de insuficiencia esqueletal a nivel mandibular puede iniciarse distracción ósea en etapa de dentición mixta o bien esperar el desarrollo completo del macizo facial, para la utilización de injertos óseos, colgajos libres microvascularizados del tipo compuesto o bien cirugía ortognática según corresponda cada caso (Figura 6 A y B).
b) Sindrome de goldenhar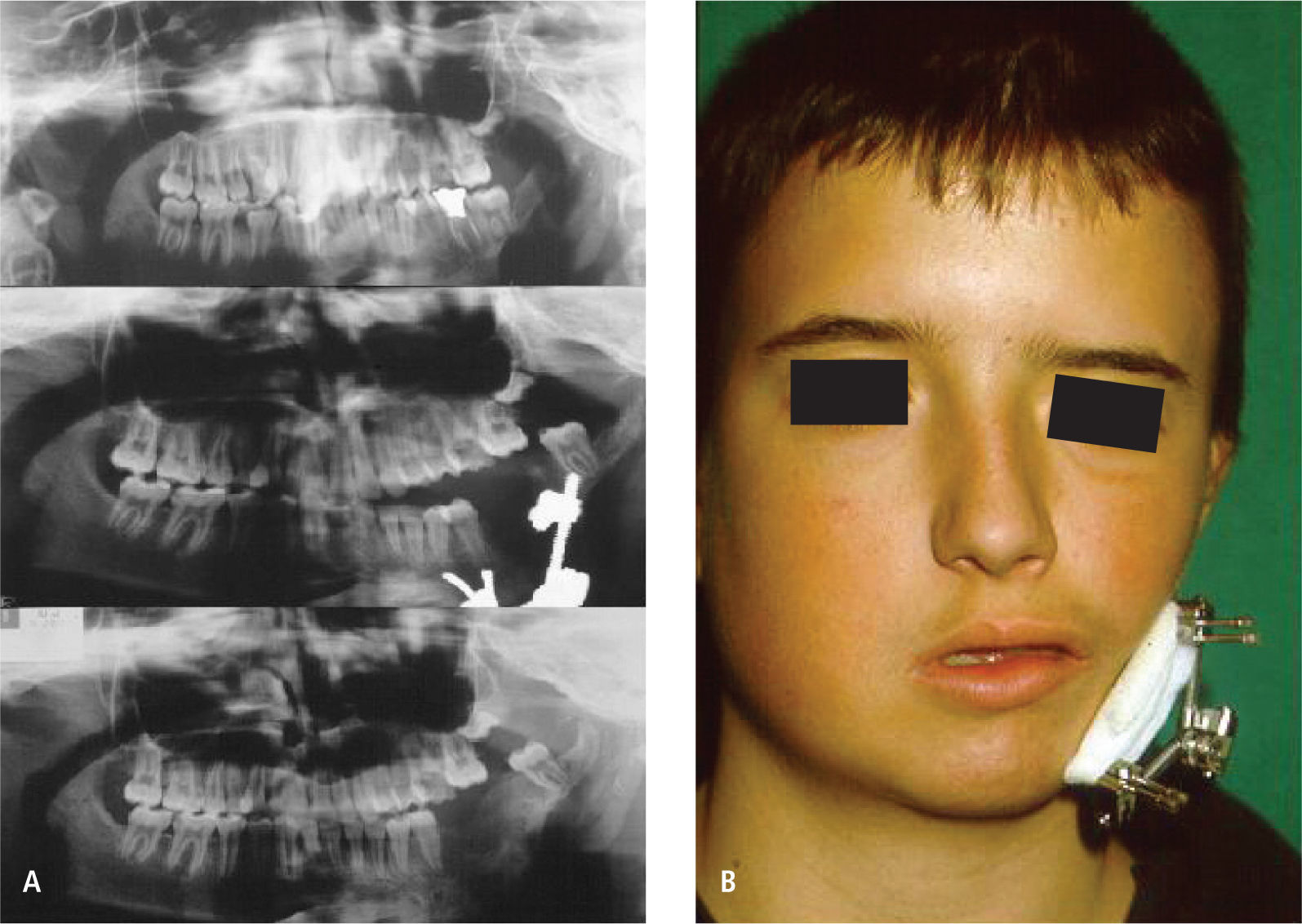
El síndrome de Goldenhar, también denominado síndrome del primer y segundo arco branquial o espectro óculo-auriculo-vertebral, es un complejo de anomalías craneofaciales y vertebrales. Fue descrito originalmente por Von Arlt, pero no fue considerado hasta 1952, cuando Goldenhar reportó tres nuevos casos de este complejo que se ha referido posteriormente con su nombre (1). En 1990, Gorlin et al (2) extendieron las especificaciones a un complejo de hechos que incluía un síndrome facioauriculoventricular, microtia, disostosis otomandibular Estos hallazgos pueden encontrarse en solitario o habitualmente asociados a microtia, hipoplasia mandibular, o malformaciones vertebrales congénitas. La incidencia es limitada y varía entre 1 caso en 45.000 a 2 en 100.000 habitantes. Actualmente se debe considerar como una malformación BILATERAL, lo que la diferenciaría de la Microsomía Hemifacial.
c) Sindrome de treacher collinsDescrito por Berry en 1889, también conocido como disostosis mandibulofacial. Se correlaciona con las fisuras faciales n° 6–7–8 de Tessier. Autosómica dominante con una incidencia de 1: 10.000 RN vivos. Anomalía simétrica y bilateral. Genéticamente correspondería a una mutación en el cromosoma 5 con su locus 5q31.33q33.3. Su etiología es desconocida. Las características del síndrome son: hendiduras palpebrales antimongolianas y colobomas del párpado inferior, hipoplasia malar, malformación del pabellón auricular y a veces del oído medio e interno, macrostomía, anomalías en la inserción de la línea pilosa, ausencia de pestañas en el tercio medial del párpado inferior. El manejo de la vía aérea en periodo neonatal es un desafío mayor dado la marcada retrusión facial. Su tratamiento es quirúrgico funcional y multidisciplinario.
d) Sindrome de nagerEl síndrome de Nager es una enfermedad poco frecuente. Las características faciales incluyen fisuras palpebrales inclinadas hacia abajo, ausencia o falta de desarrollo de la hemimandíbula inferior, malformaciones del oído medio y externo con canal auditivo atrético o estenótico, hendidura del paladar duro o blando, pestañas ausentes o más bajas, pelo del cuero cabelludo extendiéndose a la mejilla. Hay defectos en los miembros superiores que incluyen falta de desarrollo o ausencia de los pulgares y ocasionalmente, ausencia de la porción radial de la extremidad. Pueden existir otras anomalías de las extremidades como limitaciones de extensión del codo. También pueden verse afectados los dedos de los pies y piernas. Existen algunas anomalías internas incluyendo reflujo del riñón o estómago y problemas cardiacos congénitos. La gravedad del síndrome es variable. Existen aproximadamente 40 casos documentados de síndrome de Nager.
e) Sindrome de binderEl síndrome de Binder es una patología caracterizada por hipoplasia narizmaxilar, ángulo naso-frontal plano, senos frontales hipoplásicos, ausencia de la espina nasal anterior, columela corta y ángulo nasolabial agudo. El diagnóstico del síndrome de Binder es clínico y radiológico. Las características clínicas más importantes del síndrome conciernen la pirámide nasal y la oclusión dentaria. La nariz del binderiano presenta una punta aplastada y cayente a causa del defecto esquelético premaxilar y de las reducidas dimensiones horizontales del tabique nasal, las narinas aparecen de forma triangular, la columela es corta y el ángulo naso-labial es agudo. A causa de la contracción del maxilar superior debida a la atrofia premaxilar, las relaciones dento-esqueléticas resultan siempre de III clase. En los casos más serios la reducción del diámetro de las cavidades nasales en correspondencia de las narinas, en asociación a la contracción del maxilar superior, puede ser causa de distress respiratorio neonatal.
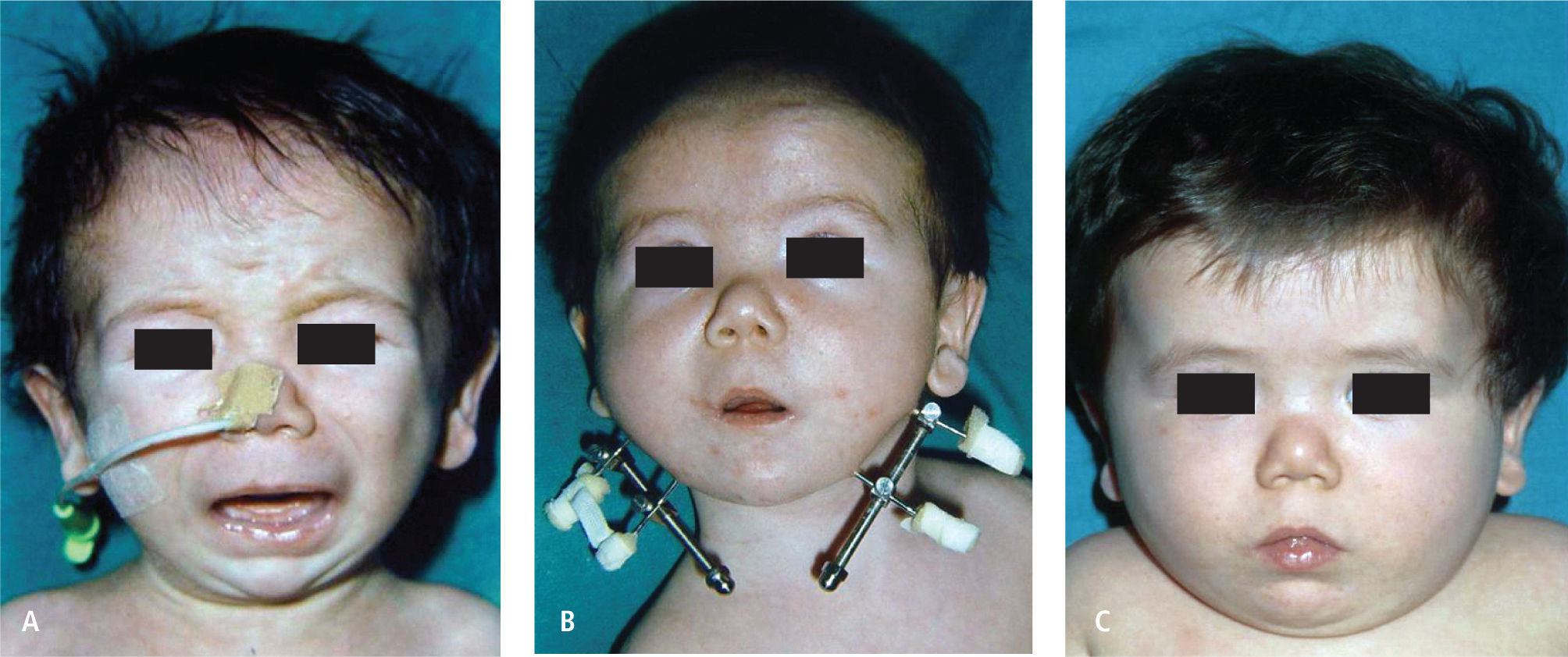
f) Secuencia De Pierre RobinEl Síndrome de Pierre Robin (SRP) fue descrito en 1923 por el estomatólogo francés quien describió la clásica triada de micrognatia, glosoptosis y obstrucción respiratoria. Esta dificultad respiratoria característica de estos pacientes sobre todo durante el periodo de recién nacidos, está dado por una hipoplasia mandibular que provoca la retroposición lingual, lo que obstruye el paso de aire. Su manejo precoz y efectivo es fundamental en la sobrevida de estos pacientes. La gran mayoría son manejados con cambios posicionales, en especial el decúbito ventral lo que permite desobstruir la retrofaringe al caer la lengua por gravedad en una posición más anterior. La monitorización continua de la saturación de O2, durante el sueño y la alimentación de estos menores determinará la efectividad de la posición. El objetivo es lograr que el niño crezca en función que su hipoplasia mandibular también lo haga. Aquellos pacientes que no logren estabilizarse o bien no logren un crecimiento ponderal de acuerdo a su edad gestacional deberán entrar a un protocolo de manejo multidiciplinario entre neonatólogos y cirujanos plásticos, donde se especifica que paciente debe ser intubado, qué paciente es candidato a una distracción mandibular bilateral y qué paciente debe ser traqueos-tomizado directamente (Figura 7).
II- Atrofia / Hipoplasiaa) Sindrome parry romberg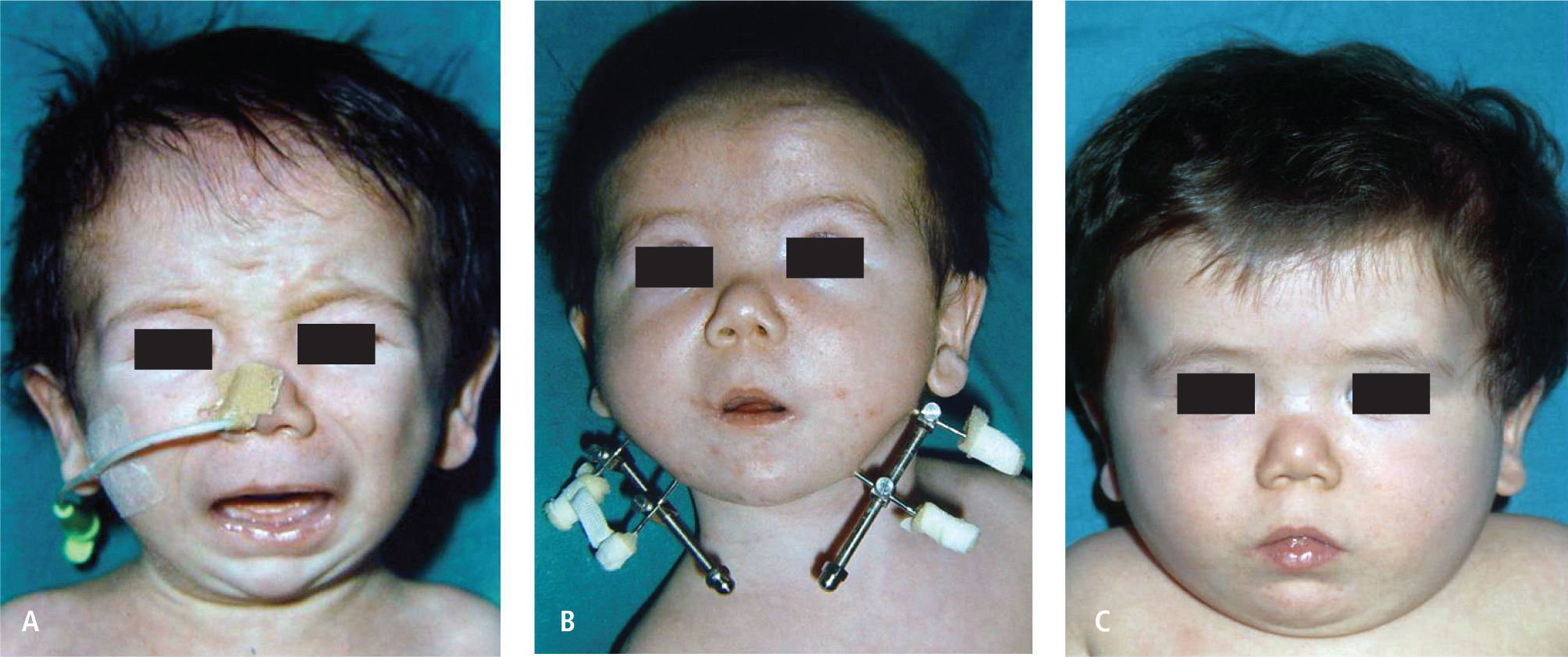
Atrofia hemifacial, entidad pocas veces vista en la práctica clínica, es una enfermedad progresiva que suele comenzar en la adolescencia o en la primera juventud. Comienza en la zona paramedial de la cara (situada a 2 ó 3 través de dedo lateralmente a la línea media) con atrofia del tejido subcutáneo graso. La piel situada por encima, los músculos faciales por debajo y en algunos casos los huesos y cartílagos de la cara, pueden estar atrofiados. Puede aparecer en la literatura con otros términos como: Enfermedad de Romberg, Síndrome de Parry-Romberg y Trofoneurosis Facial.
En las manifestaciones clínicas de la enfermedad pueden observarse con frecuencia la caída de las pestañas y una calvicie anterior; también puede haber atrofia ipsolateral de la lengua. Algunos pacientes pueden presentar epilepsia que afecta sobre todo el lado contrario y son frecuentes los procesos inflamatorios que afectan el ojo. Aunque generalmente aparece en la adolescencia o en la primera juventud, se han reportado casos en niños por diferentes autores. La piel puede presentar un color castaño claro que en casos más severos puede ser castaño oscuro; se cree que esto se debe al espesamiento de la epidermis y la atrofia de las estructuras anexas. Los músculos faciales disminuyen de tamaño, pero conservan su función, los huesos y cartílagos faciales están poco desarrollados dependiendo de la edad en que apareció la enfermedad. Ya que los huesos de la cara no alcanzan el 90% de su tamaño adulto hasta que el individuo no tiene 12 ó 13 años, de comenzar la enfermedad en la primera infancia, provocará las lesiones óseas más importantes.
Su tratamiento dependerá de la severidad de las manifestaciones clínicas, siendo de primera línea el reemplazo de aquellas estructuras atrofiadas por elementos de camuflaje tipo relleno graso con el objetivo de recobrar volumen que puede ir desde una lipoinyección, injerto dermograso hasta un colgajo libre microvascularizado.
b) Atrofia Craneofacial Secundaria RadiaciónEl uso de radioterapia en tumores craneofaciales provoca hipoplasia y falta de desarrollo de el esqueleto facial y sus partes blandas. Al tratarse de una anomalía adquirida no se profundizará en el tema.
III- Neoplasias / HiperplasiasLos tumores considerados dentro de las anomalías craneofaciales son:
- a)
Displasia fibrosa ósea
- b)
Neurofibromatosis
La craneosinostosis (CS) es una entidad que se caracteriza por el cierre precoz de una o más suturas craneales (19), lo que produce un crecimiento y desarrollo anormal del cráneo. Este concepto agrupa varios tipos según las suturas afectadas y las malformaciones asociadas. En diferentes series revisadas se calcula que de cada 10.000 RN vivos se encuentran entre 2 a 4 casos de CS, con una incidencia de 0.04% (20, 21, 22, 23, 24). En nuestro país no existe un estudio epidemiológico al respecto; pero si conocemos que el número de RN vivos a fines de la década de los noventa fue aproximadamente 250.000, cabría esperar entonces una incidencia de entre 50 y 80 casos promedio por año. Esto concuerda con el número de casos nuevos que se intervienen en un año en el Programa Nacional de Malformaciones Cráneo-Faciales del Instituto de Neurocirugía e Investigaciones Cerebrales Alfonso Asenjo, principal centro de derivación nacional en dicha patología.
Para comprender los resultados del cierre prematuro de una sutura es esencial referirse a la denominada “Ley de Wirchow” (25) según la cual, al soldarse precozmente una sutura craneal, se altera el crecimiento óseo. Este crecimiento, que normalmente tiene lugar en sentido perpendicular a dicha sutura, pasa a realizarse en forma compensatoria en otros sentidos ocasionando una alteración en el volumen o en la morfología del cráneo y la cara. Un 10% a un 20% puede desarrollar hipertensión endocraneana y presentar alteraciones clínicas y de función cerebral (vómitos, cefaleas, déficit neurológicos tales como retraso mental, ceguera, etc.).
Múltiples mecanismos han sido propuestos para explicar el cierre prematuro de las suturas craneales :
- 1.
Defecto Primario de la Bóveda
Propuesta por Virchow en 1851 (25) plantea que el defecto primario está en la propia sutura de la bóveda craneana, siendo la deformidad de la base su consecuencia. Para el autor se trataba de un proceso inflamatorio, probablemente secundario a la sífilis que se desarrollaba en las meninges y afectaba los huesos del cráneo.
- 2.
Intraútero
En 1907 Thoma (26) formula la hipótesis de una presión externa que produce el cierre patológico de alguna sutura durante la etapa fetal. Para esto se requería de la presencia de oligohidroamnios en un útero gravídico.
- 3.
Defecto Primario de la Base
En 1959 Moss (27) plantea que el defecto primario es debido a cambios en la base del cráneo. Él enfatiza que la duramadre está íntimamente adherida a la base craneal en cinco puntos fundamentales: apófisis crista gallis, alas menores del esfenoides y crestas petrosas, donde hay tractos fibrosos durales que se dirigen a la bóveda en la misma dirección que las suturas craneales. Estos tractos transmitirían las fuerzas mecánicas recíprocas entre la base y la bóveda dirigiendo las líneas de crecimiento encefálico en direcciones específicas. Sin tales tractos el neurocráneo tendría una forma completamente esférica.
- 4.
Alteración primaria del Mesénquima.
Planteada inicialmente por Park y Powers (28), propone que la causa primaria es un crecimiento defectuoso del mesénquima en el cual se forma el hueso. Los huesos formados en este tejido son más pequeños que los normales y hacen contacto uno con el otro muy pronto. Como se conoce, los márgenes de los huesos del cráneo se mantienen separados por el crecimiento intersticial de tejido mesenquimatoso. Al existir un defecto en el plasma germinal, la capacidad de crecimiento normal del mesénquima está disminuida o abolida, sin desarrollarse el exudado que tiene la capacidad de resistir la osificación. Esto explica la sinostosis prematura, ya que tan pronto los huesos toman contacto se fusionan. Las lagunas de mesénquima han sido demostradas experimentalmente (29).
- 5.
Hereditaria
Factores hereditarios han sido citados por muchos investigadores (18, 19, 20). Estos sugieren alteraciones en los genes que se van transmitiendo. En algunos tipos como el Síndrome de Crouzon (CS) y el de Apert estos factores han sido determinados por mutaciones del cromosoma 10, locus FGFR2 (21, 22).
La clasificación más utilizada por ser considerada útil y práctica corresponde a la de los autores David y Poswillo (23) que las divide en dos grandes grupos:
- A.
-No Sindromáticas O Simples
- 1.
-Escafocefalia
- 2.
-Trigonocefalia
- 3.
-Plagiocefalia
- 4.
-Oxicefalia
- 5.
-Braquicefalia
- 1.
- B.
-Sindromáticas O Complejas
- 1.
-Crouzon
- 2.
-Apert
- 3.
-Carpenter
- 4.
-Chotzen
- 5.
-Pfeiffer
- 6.
-Otros Síndromes
- 1.
La CS se caracteriza por un cráneo morfológicamente anormal. En las fases iniciales del proceso el desarrollo cerebral no se altera ya que el cráneo puede crecer todavía en los diámetros no perpendiculares a la sutura afectada, según la “Ley de Wirchow” (10).
Más adelante, cuando la consolidación es completa, el crecimiento encefálico puede llegar a originar hipertensión endocraneana. Sin embargo es evidente por la alta frecuencia de presentación de las CS de bajo riesgo en nuestro medio, que los cuadros hipertensivos son muy pocos. La dismorfia craneal es de aparición precoz, precediendo en algún tiempo a las manifestaciones neurológicas que se pueden originar. Esta deformidad está directamente relacionada con la sutura afectada y, según la conformación que adopte, recibirá diferentes denominaciones que veremos a continuación:
- 1.
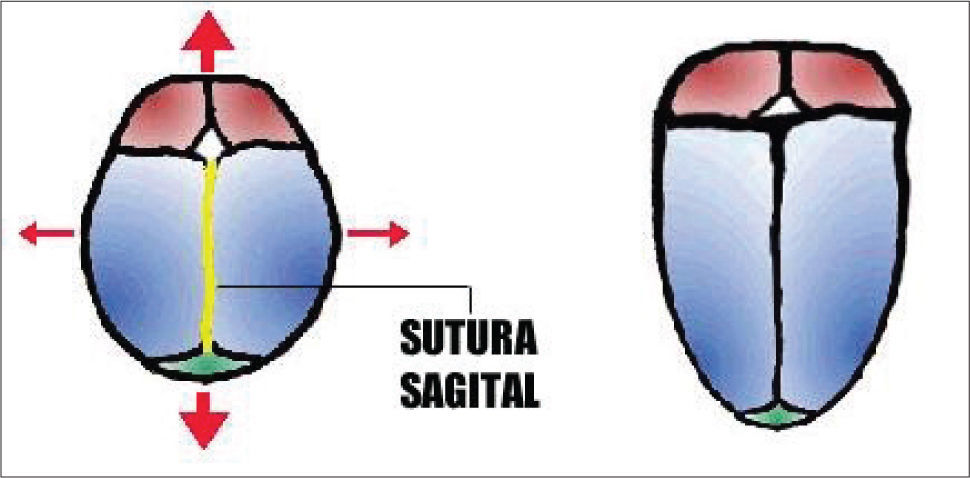
Escafocefalia: Cierre precoz y exclusivo de la sutura sagital que separa a los huesos parietales, lleva al crecimiento del cráneo paralelo a la sutura cerrada y a la imposibilidad de crecimiento transversal. El resultado es una cabeza alargada en sentido anteroposterior que recuerda a un barco volcado (escafo: término griego que significa barco). De buen pronóstico ya que no produce hipertensión intracraneana y es, por tanto, un problema esencialmente estético. Corresponde por sí sola prácticamente al 50% de las formas de CS (Figura 8).
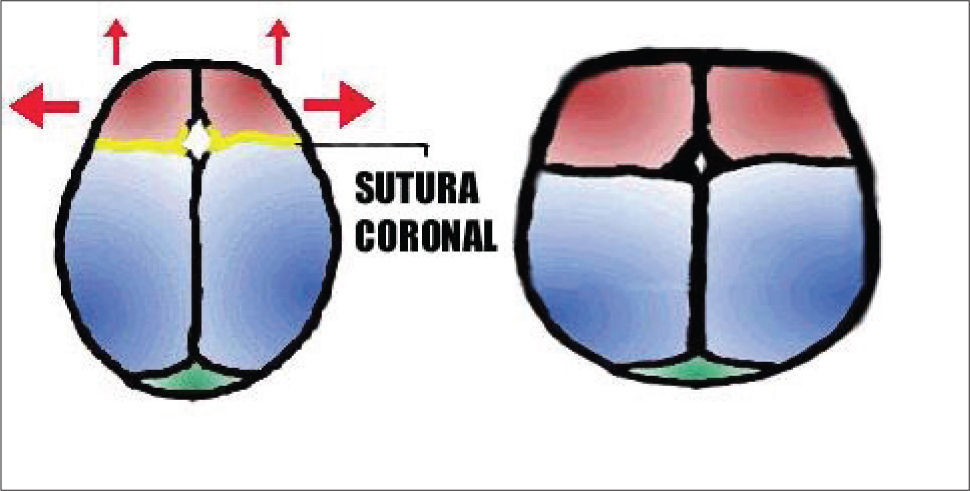
- 2.
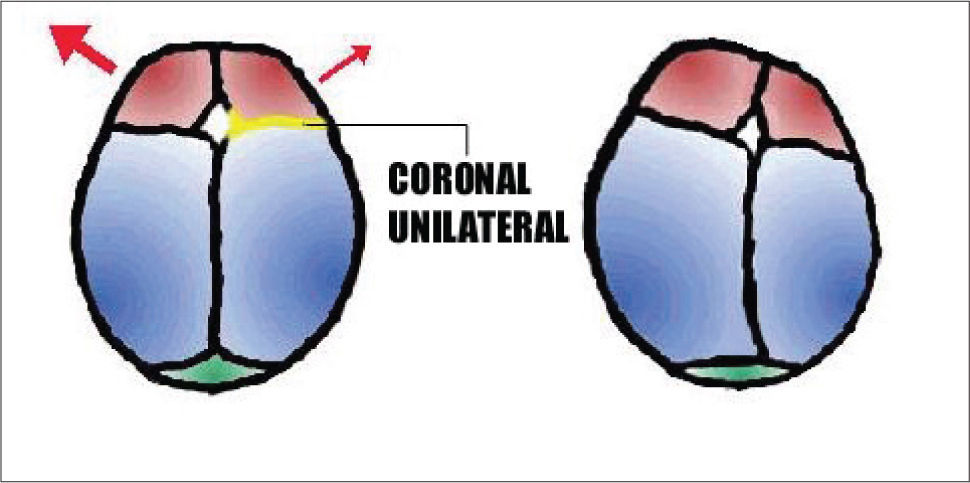
Braquicefalia: Cierre precoz y bilateral del complejo suturario coronal, constituido a su vez por dos componentes; el primero fronto-temporal cuyo compromiso originará un cráneo corto y ancho, el segundo etmoido- esfenoidal del cual resulta al comprometerse la hipoplasia del tercio medio facial. Si el cierre prematuro se limita a sólo una sutura coronal el resultado es la PLAGIOCEFALIA. Cuando el compromiso es bilateral y de ambos componentes ocasiona exoftalmo, hipertelorismo y retrusión del tercio medio facial. En el segundo, la deformidad es asimétrica con aplanamiento del lado afectado e hipoplasia de la órbita correspondiente, más en algunos casos dismorfia facial evidente (Figura 9 y 10).
- 3.
Trigonocefalia: (cráneo en cuña) Resulta del cierre prematuro de la sutura frontal o metópica. La frente es estrecha y prominente en su línea media con una forma triangular y se aprecia hipotelorismo. Su interés se fundamenta en el aspecto estético y su asociación a malformaciones neurológicas intracraneales.
- 4.
Oxicefalia: Cierre precoz de todas las suturas del cráneo. CS de diagnóstico tardío, que según muchos autores no existiría con clínica florida antes de los 3 años de vida. Tiene dos formas de presentación, la clásica o asimétrica característica del continente africano y la forma simétrica de crecimiento “armónico” que predomina en nuestro medio entre los 2 y 3 años. Los antecedentes son de un cierre prematuro de fontanelas, en los cuales existe un cráneo armónicamente pequeño y cuyo diagnóstico diferencial se plantea con la microcefalia resolviéndose el problema al constatar impresiones digitiformes en las radiografías de cráneo y fundamentalmente conflicto de espacio en la TAC (31).
- 5.
Mixtas o complejas: Resultan del cierre precoz de dos o más suturas y su combinación. La TURRICEFALIA que se produce por el cierre completo de la sutura coronal con un crecimiento del cráneo hacia arriba recordando la forma final de una torre. La ACROCEFALIA cuyo crecimiento es esférico. Ambas originan con frecuencia no despreciable hipertensión endocraneana y sus consecuencias como lo son: retraso mental y trastornos visuales producto del exorbitismo y por compresión del nervio óptico.



Además de estas variedades que constituyen CS primarias simples existen las complejas, así denominadas por acompañarse de otras alteraciones. De estas se han descrito más de 67 síndromes genéticos (21, 22, 30, 32, 33) de los cuales citaremos los más frecuentes:
- •
Enfermedad de Crouzon: También conocida como sinostosis craneofacial. Comúnmente es hereditaria en forma dominante, aunque algunos casos se producen en forma esporádica, sin historia familiar. Cursa con braquicefalia, nariz en gancho, hipoplasia maxilar, labio superior corto e inferior saliente, hipertelorismo, exoftalmos y estrabismo divergente.
- •
Síndrome de Apert: Llamado acrocefalosindactilia. Se presenta con braquicefalia asociada a sindactilia de los pies y las manos, atresia de coanas, megalocórnea, estrabismo, hipoplasia orbitaria y otras malformaciones. Se piensa que puede tener un carácter de transmisión autosómica dominante.
La hipertensión endocraneana provocaría anosmia, ceguera, sordera y oftalmoplejias (34). Los casos descompensados por el aumento de la presión intracraneal pueden presentarse clínicamente con cefaleas, retraso mental, exoftalmos, crisis epilépticas, vómitos, irritabilidad o lesión de un nervio craneal acompañado de defecto motor.
También se han descrito múltiples defectos asociados, entre lo que tenemos (35, 36, 37):
- Alteraciones craneofaciales o encefálicas: Hipoplasia del maxilar, prognatismo, platisbasia, atresia de coanas, paladar hendido, malformación de Arnold-Chiari, dismorfia de pabellón auricular, hipoplasia del cuerpo calloso, hidrocefalia, holoprosencefalia.
- Alteraciones de las extremidades: Sindactilia, polidactilia, braquidactilia, aplasia del radio, pulgar en delta.
- Otras alteraciones: Porfinurias, criptorquídeas, obesidad, cardiopatías congénitas, hipogonadismo, espina bífida.
Un cráneo deformado al nacer no debe interpretarse como una CS. Existe un sin número de malformaciones asociadas como la posición fetal in útero, utilización de forceps durante el expulsivo, la postura de descanso que el menor desarrolle durante la lactancia. La palpación puede no revelar anomalías al nivel de las suturas o bien palpar fontanelas abiertas, sin embargo es la Rx la que finalmente nos determinará la presencia o no de las suturas craneales y las disposiciones características de cada sindrome. Las malformaciones posicionales desaparecen espontáneamente.
Aparte de la Oxicefalia, por ser una CS simétrica, todas las otras CS son pesquisables al momento de nacer.
El primer síntoma es la forma del cráneo. A la palpación se siente muchas veces un puente que une una o más suturas. La medición del diámetro de la bóveda craneal, el examen neurooftalmológico y la Tomografía también son fundamentales en el acercamiento al paciente con CS.
El Avance Fronto-Orbitario (AVFO) con remodelación frontal es el tratamiento de elección de la mayoría de las CS. En la actualidad y durante los últimos dos años en el INC Asenjo de Santiago basado en cirugías mínimamente invasivas se ha desarrollado la remodelación en base a la distracción ósea utilizando alambres, los cuales basados en su fuerza tensil logran la remodelación y expansión necesaria sin grandes movilizaciones óseas, lo que acorta la evolución post operatoria disminuyendo los riesgos y las necesidades de transfusión sanguínea. Dado que es una experiencia preliminar nos avocaremos al sistema tradicional.
El AVFO considera dos aspectos fundamentales como son:
- 1.
Restaurar la anatomía normal de la frente y la bóveda craneana.
- 2.
Permitir la expansión del cerebro y aprovechar la fuerza con que se produce.
La frente es dividida en dos partes: (1) la barra fronto-orbitaria y (2) la porción vertical del frontal que asciende formando una curva suave hacia ambos parietales.
Estas dos partes son tratadas en forma separada. La barra fronto-orbitaria se remodela y se fija avanzándola en posición ventajosa, para luego ajustar sobre ésta en forma concordante la porción superior. El método de fijación y de estabilización de las plaquetas dependerá de la realidad de cada servicio (alambres, placas-tornillos, vicryl, seda etc.).
En casos de CS sindromáticas, en la actualidad lo que estamos utilizando es la distracción centrofrontocraneofacial, para lo cual utilizamos una osteotomía en monoblock, con lo cual logramos avanzar en forma progresiva y pausada el macizo facial completo en un solo tiempo apoyado en los distractores diseñados por Arnaud.
La velocidad de crecimiento cerebral de los lactantes es extremadamente rápida, llegando a doblar su tamaño en los primeros seis meses. Esto nos permite que al reparar el defecto de la CS, se mantengan los nuevos vectores de crecimiento otorgados por la remodelación, utilizando el empuje del cerebro al ir creciendo.
ResumenLa CS afecta 2 a 3 niños cada 10.000 RN vivos.
El incremento de la presión intracraneal en forma paulatina, así como los desórdenes oftalmológicos y de conducta justifican una conducta quirúrgica precoz.
El AVFO resuelve en la mayoría de los casos el problema funcional y estético a la vez. La remodelación debe efectuarse durante su primer año de vida.
El autor declara no tener conflictos de interés, en relación a este artículo.