







La atrofia muscular espinal (AME) es la primera causa genética de mortalidad en lactantes. La severidad de las manifestaciones clínicas es un continuo, con tres principales subtipos en pediatría: AME1 que se presenta en recién nacidos y no logran sentarse, AME2 en niños que no logran caminar pero sí sentarse y AME3 que logran caminar. La complicación más seria es la insuficiencia respiratoria. El enfoque del manejo respiratorio es preventivo, con toma de decisiones anticipadas por parte de los cuidadores, que incluye optimizar el manejo de la tos, evitar la deformación de la caja torácica y permitir un desarrollo pulmonar adecuado, tratar la hipoventilación, manejar oportunamente las infecciones respiratorias, el trastorno de deglución, el reflujo gastroesofágico y la malnutrición. A las puertas del desarrollo de tratamientos específicos modificadores de la enfermedad, mediante oligonucleótidos antisentido o vectores genéticos entre otros, los cuidados en AME con enfoque multidisciplinario nos imponen nuevos desafíos donde los cuidados respiratorios deberían estar de acuerdo a lo sugerido en consensos de estándar de manejo, optimizando sus condiciones globales a la espera de tratamientos más específicos.
Spinal muscular atrophy (SMA) is the first inherited cause of mortality in infants. The clinical severity is a continuos with three subtypes in children: SMA1 in newborns non sitters, SMA2 sitters and SMA3 walkers. Respiratory insufficiency is the most severe complication. The respiratory care is preventive, with parental decisions being relevant: it includes cough assistance, prevent chest deformity and lung underdevelopment, treat aggressively respiratory infections, hypoventilation, swallow problems, gastroesophageal reflux and malnutrition. In the perspectives of the new therapeutics advances, with nonsense oligonucleotids or gen therapy, we need to optimize treatments with standard of care in a multidisciplinary approach.
La atrofia muscular espinal (AME) ligada al cromosoma 5q13 tiene una incidencia de 1/6000-10000 nacimientos y es la primera causa genética de mortalidad en lactantes. Esta enfermedad es consecuencia de una delección o mutación homocigota del gen de sobrevida de la motoneurona (SMN1) que ocasiona el daño y muerte de las motoneuronas alfas que se originan en la médula espinal y troncoencéfalo 1,2. Las manifestaciones clínicas tienen un amplio espectro de gravedad en relación al grado de debilidad muscular y compromete principalmente las extremidades y en forma variable, la musculatura respiratoria y bulbar, con nivel intelectual normal. Si bien se reconoce que la severidad de las manifestaciones clínicas en AME es un continuo, se han definido tres principales subtipos en la población pediátrica: AME tipo 1 que se presenta en recién nacidos y lactantes incapaces de sentarse, AME tipo 2 en niños que no logran caminar pero sí sentarse y AME tipo 3 en niños que logran caminar de manera independiente. En los seres humanos existen 2 genes SMN casi idénticos en el cromosoma 5q13, uno es SMN1 telomérico cuya delección es responsable de la AME y otro SMN2 centromérico, que difieren solo en un cambio nucleotídico en la secuencia codificante en el exón 7, que afecta el splicing o empalme, provocando la exclusión del exón 7 en la transcripción de la proteína del gen SMN2. Se produce entonces una proteína truncada que se degrada rápidamente en un 80% a 90% de las veces. El 10% de proteína SMN completa que produce la trascripción del gen SMN2 no alcanza a compensar la ausencia de proteína producida por la delección de SMN1. Sin embargo, el gen SMN2 es un modificador de la gravedad de la enfermedad y se encuentra en un número variable de copias en los individuos. El número de copias de SMN2 está inversamente relacionado con la gravedad de la enfermedad, siendo un importante, pero no el único factor modificador de la expresión fenotípica 2,3,4.
Los pacientes con AME tipo 1 representan cerca del 50% de todos los pacientes con AME. En este tipo de pacientes, la historia natural de la enfermedad ha mostrado que fallecen antes de los 24 meses de edad por insuficiencia respiratoria. Esta realidad se ha modificado en los últimos años debido al aumento de los cuidados clínicos preventivos, asociados a una mayor comprensión y conocimiento de la AME. El pronóstico vital de estos pacientes se ha modificado significativamente con la incorporación de apoyos nutricionales y respiratorios propuesto en la declaración de consenso de expertos sobre los cuidados estándar en pacientes con AME 5,6. Un porcentaje creciente está viviendo más allá de los 2 años 7.
En la actualidad nos encontramos a las puertas del desarrollo de tratamientos específicos modificadores de la enfermedad ya que en el último decenio se ha producido un incremento explosivo en el desarrollo de nuevas moléculas y estrategias terapéuticas específicas para la AME, respaldadas por ensayos clínicos prometedores que se están realizando en muchos países del mundo. Es bajo esta perspectiva que se ha querido realizar esta revisión, destacando la importancia de los cuidados respiratorios en atrofia muscular espinal a la luz de los desafíos impuestos frente la emergencia de nuevas terapias, ya que debemos redoblar los esfuerzos por mantenernos atentos en perfeccionar las atenciones en salud de estos pacientes y así optimizar sus condiciones globales a la espera de tratamientos más específicos 8,9.
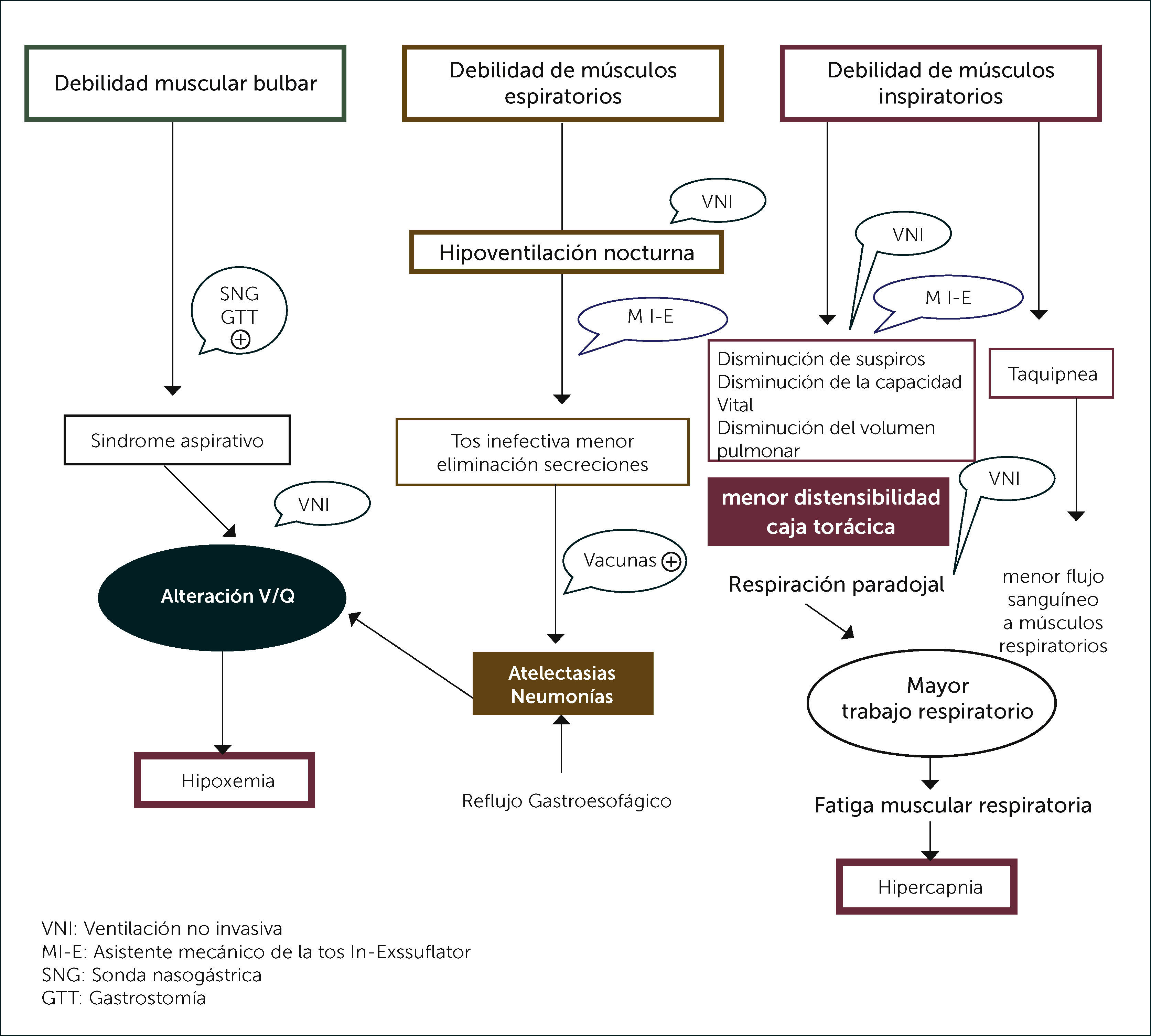
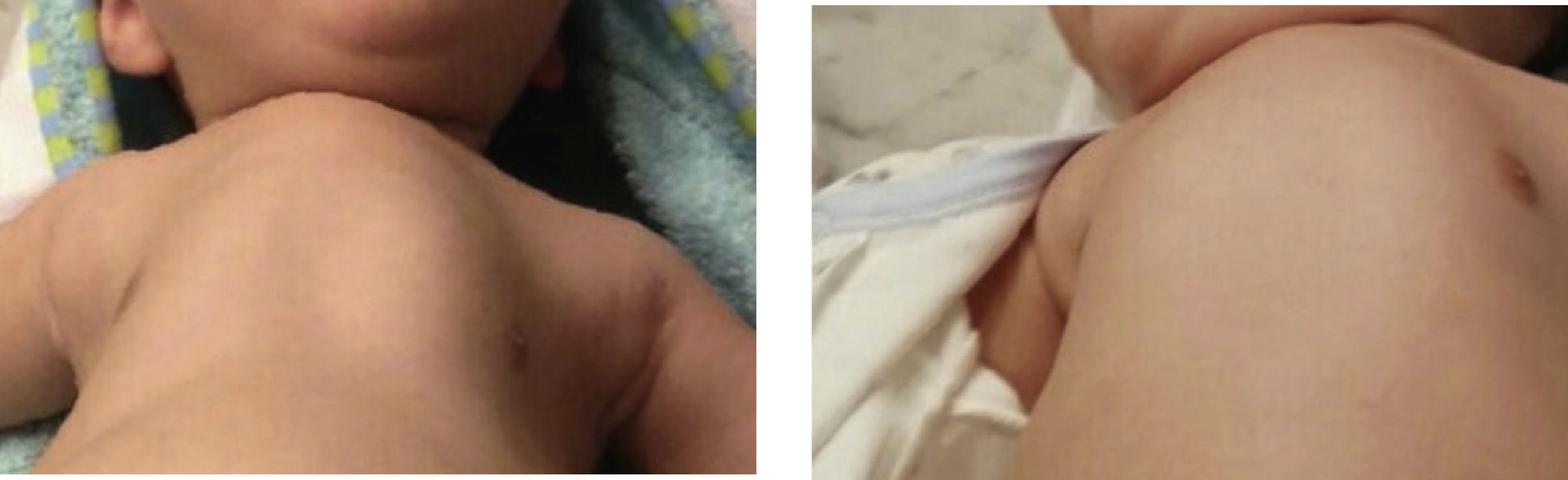
FISIOPATOLOGÍA DEL COMPROMISO RESPIRATORIOLa complicación más seria en AME es la insuficiencia respiratoria que es de causa multifactorial y con mecanismos relacionados entre sí; conocerlos permitirá implementar intervenciones específicas para cada enfermo en particular. En estos pacientes existe compromiso de todos los grupos musculares respiratorios importantes en forma variable, dependiendo del tipo de AME y del estado de la enfermedad: músculos inspiratorios, espiratorios y bulbares, con un diafragma relativamente más fuerte. Debido a este desbalance entre contractilidad diafragmática y distensibilidad de la caja torácica, los niños con AME 1 y 2 desarrollarán un tórax acampanado y una depresión esternal o pectus escavatum5,6. La probabilidad de presentar insuficiencia respiratoria en AME 1 en inevitable, frecuente en AME 2 e infrecuente en AME 3 (Tabla 1). La debilidad de los músculos inspiratorios no permite alcanzar una inspiración máxima, lo que disminuye la generación de suspiros. Esto favorece el desarrollo de microatelectasias que reduce la capacidad vital (CV) al disminuir la distensibilidad pulmonar y torácica. Con el tiempo, los tendones, ligamentos y articulaciones costales pueden anquilosarse debido a la fibrosis y acortamiento de las fibras musculares. La respiración es rápida, superficial, sin suspiros y puede ser paradojal, aumentando el trabajo respiratorio, la ventilación de espacio muerto y el tiempo inspiratorio. El desbalance entre la carga de trabajo de los músculos débiles y su capacidad favorece la fatiga muscular y la insuficiencia respiratoria, por la insuficiente actividad diafragmática en posición supina o por disfunción muscular generalizada, la que inicialmente ocurre durante el sueño. Los músculos espiratorios, predominantemente abdominales e intercostales, son importantes para lograr una tos efectiva y la eliminación de secre- ciones. Su compromiso favorece las complicaciones como el colapso alveolar y el desarrollo de hipoxemia secundaria, adquiriendo mayor relevancia en las infecciones respiratorias y la anestesia general. El compromiso bulbar, especialmente importante en AME 1 y variable en AME 2, es causa de la alteración de la deglución de saliva y alimentos, favoreciendo la aspiración aguda y crónica. Las alteraciones respiratorias del sueño son de origen central, obstructivas o de origen mixtas. La hipoventilación en el sueño con movimientos oculares rápidos (REM) ocurre por disminución del volumen corriente, especialmente en pacientes con disfunción diafragmática severa; esto se relaciona con la inhibición de la actividad de los músculos intercostales y accesorios. Con el tiempo la hipoventilación favorece los eventos hipóxicos, la retención de bicarbonato y la depresión del centro respiratorio, creando un círculo vicioso. Los eventos obstructivos ocurren durante la inspiración, ya que se genera una presión negativa que actúa sobre debilidad de la faringe y la laringe produciendo el colapso de la vía aérea superior. Como estos eventos aparecen inicialmente durante el sueño se recomienda investigarlos activamente 10. La escoliosis, frecuente en AME 2, altera la mecánica torácica y el funcionamiento muscular, contribuyendo a la limitación restrictiva y al desarrollo de atelectasias 11. Estudios recientes han demostrado que AME es una enfermedad neuromuscular bastante estable en su compromiso, con una disminución lenta de la capacidad vital y de otros parámetros, y que su progresión se relaciona a la insuficiente ganancia de fuerza y masa muscular para alcanzar las demandas asociadas al crecimiento y desarrollo normal del niño 12. Este concepto refuerza la importancia de los cuidados respiratorios preventivos en AME.
CLASIFICACIÓN DE LA ATROFIA MUSCULAR ESPINAL Y PRONÓSTICO RESPIRATORIO
| AME | Insuficiencia Respiratoria | Máxima función adquirida | Neumonías | Pronóstico | Clasificación | Soporte ventilatorio |
|---|---|---|---|---|---|---|
| 1 | Todos antes de los 2 años | No se sientan | Todos | <2 años | 1a inicio neonatal 1b inicio posterior, nunca controlan cabeza 1c controlan cabeza | Todos |
| 2 | 40% antes de los 2 años | Se sientan, pero nunca se paran | 25% en la niñez | Sobreviven hasta la adultez | Depende del nivel de función | Variable |
| 3 | > 18 meses | Se paran y caminan | Raro en la niñez | Lenta, sobreviven hasta la adultez | 3a inicio de debilidad antes de los 3 años 3b después de los 3 años | Variable, en la adolescencia |
| 4 | 10-30 años | Se paran y caminan | Raro | Sobreviven hasta la adultez | No |
Las recomendaciones para la evaluación de la función respiratoria en pacientes con AME se resumen en la Tabla 2. En niños pequeños, especialmente en aquellos que no se sientan o son muy débiles para realizar pruebas de función pulmonar la evaluación debe ser indirecta. Es importante vigilar la efectividad de la tos, la frecuencia respiratoria, la respiración paradojal y el intercambio gaseoso. En los pacientes con AME la tos se afecta precozmente, por lo que en lactantes y niños pequeños debe ser evaluada clínicamente y sobre los 12 años a través del pico flujo de tos (PFT) mediante un flujómetro portátil o con el neumotacómetro utilizado para la realización de la espirometría, con una boquilla o interfase nasal en caso de debilidad de los músculos bucales. Para ello es necesario realizar una inspiración inicial mayor a 85-90% de la capacidad de insuflación máxima (CIM) y una presión toracoabdominal superior a 100cmH2O. Los factores que influyen en la eficacia de la tos son la CV, la CIM y la presión inspiratoria máxima (PIMax), siendo menos importante la presión espiratoria máxima (PEMax). Valores de PFT menores a 270L/min han demostrado un mayor riesgo de complicaciones respiratorias. Cuando el PFT es menor a 160 la tos es inefectiva 13,14. La CV en posición erguida se puede medir en la mayoría de niños desde los 6 años y puede estar disminuida; una diferencia de 20% con la CV en decúbito supino permite evidenciar la disfunción diafragmática. Una CV <1.1 litro se asocia a un riesgo mayor de complicaciones durante las infecciones respiratorias 15,16. En pacientes con escoliosis se recomienda la estimación de la talla a partir de la medición de la envergadura o de la longitud cubital. La evaluación de la fuerza de los músculos respiratorios se realiza habitualmente a través de la medición de la PIMax y PEMax mediante un transductor de presión que el paciente coloca en su boca cuando éste realiza esfuerzo inspiratorio máximo a partir de espiración completa y esfuerzo espiratorio máximo después de inspiración completa; la maniobra de PIMax se realiza desde volumen residual, mientras que la de PEMax desde capacidad pulmonar total. Una PIMax disminuida refleja debilidad de los músculos inspiratorios, principalmente el diafragma y la PEMax los músculos espiratorios, incluidos los abdominales. La PIMax es una maniobra simple, sin embargo tiene algunos problemas: requiere coordinación entre el técnico y el paciente, mantener por un mínimo tiempo el esfuerzo máximo, aprendizaje, en ocasiones participa la musculatura facial, se requieren de 5 a 9 maniobras válidas y cuando hay problemas de la vía aérea superior pueden provocar su colapso inspiratorio que dificulta la transmisión de la presión alveolar a la boca. Una técnica complementaria a la PIMax es la medición de la presión inspiratoria nasal máxima (SNIP o sniff nasal inspiratory pressure) cuyos valores de referencia han sido validados para niños sobre 6 años. Es una maniobra no invasiva y dinámica para la determinación de la fuerza de los músculos inspiratorios en la fosa nasal que consiste en realizar una inhalación forzada, generalmente desde capacidad residual funcional y medición de la presión generada. Su única dificultad es cuando existe obstrucción nasal; no requiere coordinación entre técnico y sujeto evaluado, tampoco mantener el esfuerzo en el tiempo, no participa la musculatura facial y no se altera frente a problemas de la vía aérea superior 10,16. La polisomnografía evalúa la fragmentación del sueño y la hipoxia e hipercapnia recurrente que deterioran los mecanismos de reacción frente a la apnea al aumentar la tolerancia de los quimiorreceptores y pueden elevar el umbral de la reacción de despertar, lo que agrega una hipoventilación central secundaria 11.
RECOMENDACIONES PARA EL CUIDADO RESPIRATORIO DE LA ATROFIA ESPINAL TIPO 2 Y 3
| MAYORES DE 6 AÑOS |
| CVF sentado, PFT, PIM, PEM, SNIP anual si CVF > 60% y cada 6 meses si es < 60% |
| A CUALQUIER EDAD |
| Vacunas neumococo, influenza anual |
| Descartar: trastornos respiratorios del sueño, tos débil, síntomas de disfunción bulbar (atoros, disfagia, salivación), RGE, infecciones respiratorias recurrentes |
| Evaluar saturación de oxígeno, tos, respiración paradojal, forma y tamaño de la caja torácica |
| Saturometría nocturna |
| Gases arteriales en infecciones respiratorias agudas e insuficiencia respiratoria, sat < 95%, CVF < 40% |
| Radiografía de tórax al menos cada 2 años |
| TcPCO2 si está disponible en caso de sospecha de hiperventilación nocturna |
| Polisomnografía en caso de sospecha de trastornos respiratorios del sueño: SAHOS, hipoventilación |
| Evaluación por cirujano de columna |
| Evaluación de la deglución frente a la sospecha de alteración: fonoaudiología, videodeglución |
CVF: capacidad vital forzada: PFT: pico flujo de tos; PIM:presión inspiratpria máxima; PEM: presión espiratoria máxima;
SNIP: presión inspiratoria nasal sniff; RGE: reflujo gastroesofágico; SAHOS: síndrome de apnea hipopnea del sueño.
Adaptado de Sansone, Racca F, Ottonello G, et al. on behalf of the Italian SMA Family Association, 1st Italian SMA Family Association Consensus Meeting:, Neuromuscular Disorders 2015; 979-989.
El enfoque del manejo en AME es principalmente preventivo, donde la educación de los cuidadores en fundamental; Ellos deben comprender el objetivo de los tratamientos y además tomar decisiones anticipadas, con un apoyo multidisciplinario de neurólogo, pediatra neumólogo, sicólogo, fisiatra y nutriólogo que es esencial. Las claves en los cuidados respiratorios preventivos incluyen mejorar el manejo de la tos y favorecer la eliminación de secreciones respiratorias, evitar la deformación de la caja torácica, la respiración paradojal y el tórax en campana con el fin de permitir el desarrollo pulmonar adecuado, pesquisar y tratar la hipoventilación durante el sueño y manejar oportunamente las infecciones respiratorias recurrentes que aumentarán aún más la debilidad muscular. Es necesario diagnosticar y tratar la alteración de la deglución y el reflujo gastroesofágico, ya que la aspiración pulmonar se verá agravada por la tos inefectiva (Figura 1). La desnutrición puede aumentar la debilidad. La escoliosis progresiva, la distensión abdominal producida por fecalomas y la obesidad alterarán la mecánica respiratoria, por lo cual deben ser monitorizadas y tratadas oportunamente. La obesidad frecuentemente observada en la adolescencia en niños con AME 2 y 3 favorece los trastornos respiratorios del sueño. No existe correlación directa entre la alteración pulmonar y la necesidad de asistencia ventilatoria. La evidencia sobre el mejor manejo es escasa y está basada principalmente en series de casos y en consensos de expertos de estándar de manejo 5,6,17. Las evaluaciones deben ser periódicas, más frecuentes en AME 1 y cada 3 a 6 meses en pacientes con AME 2 y 3 con el fin de diagnosticar los problemas en forma precoz. Se debe administrar las vacunas necesarias, incluidas neumococo y anualmente influenza. Se ha sugerido considerar en algunos casos el uso de anticuerpos monoclonales contra el virus respiratorio sincicial. El favorecer la tos y eliminación de secreciones en estos pacientes es crítico. La asistencia de la tos debe comenzar en forma paralela al inicio del apoyo ventilatorio no invasivo. Esto puede lograrse también mediante la asistencia manual con ambú, air stacking o idealmente con el asistente mecánico insufflator-exsufflator (MI-E) diariamente para evitar la formación de tapones mucosos y microatelectasias. En AME 2 y 3 maniobras como la respiración glosofaringea ayudan a este proceso 18–20. En AME 1 y 2 la debilidad muscular respiratoria es mayor para los músculos intercostales, de manera que el diafragma es el principal músculo de la ventilación. El tórax en los primeros años de vida es además muy distensible, lo que favorece su deformación; la expansión es necesaria para mantener su distensibilidad y promover el crecimiento pulmonar (Figura 2). La asistencia mecánica de la tos a través de una interfase oronasal puede ser utilizada desde lactantes, ya sea en forma rutinaria, diariamente para la expansión pulmonar y su adecuado desarrollo y especialmente durante las infecciones respiratorias, aumentándola si la saturación de oxígeno cae bajo 95%. La cooperación se obtiene desde los 2 años, sin embargo, en menores de 12 meses es necesario ajustarlo a los esfuerzos inspiratorios y espiratorios. Puede hacerse compresión abdominal durante la fase de exsuflación. Las presiones efectivas son entre ±30 a ±60cm H20. El objetivo es lograr una expansión rígida y completa y luego vaciar el tórax. En algunos pacientes será necesaria la aspiración, especialmente en AME 1, donde la alteración del reflejo de deglución favorece la acumulación de saliva 19,20.


La ventilación mecánica no invasiva (VMN) puede iniciarse cuando hay evidencia de hipoventilación nocturna cuyos síntomas son despertares, cefalea matinal, fatiga, somnolencia diurna y dificultad en la concentración y trabajo escolar. Esto puede ser evaluado -aunque no siempre es necesario- mediante polisomnografía, o en su defecto poligrafía u oximetría continua nocturna en forma ambulatoria, especialmente útil en pacientes con AME 2 y 3. En ocasiones los gases matinales pueden ayudar, aunque son de baja sensibilidad, además de ser molestos para el niño. La VMN es necesaria además cuando existe dependencia para gatillar un flujo de aire adecuado, cuando hay descompensaciones con atelectasias que requieren la administración de oxígeno, para lograr una extubación exitosa, en anticipación a la descompensación por infección respiratoria, cuando existe dependencia de ella y para dar de alta al domicilio con cuidados paliativos 21–25. Los consensos internacionales sugieren que también debería iniciarse si existe tos inefectiva 4,5 (Tabla 3).
OBJETIVOS DE LA VENTILACIÓN MECÁNICA NO INVASIVA EN ATROFIA MUSCULAR ESPINAL
| Control de la hipoventilación nocturna |
| Control de la hipoventilación diurna |
| Apoyo en las infecciones respiratorias agudas |
| Uso perioperatorio de cirugías: escoliosis, gastrostomía |
| Favorecer el tamaño de la caja torácica y evitar deformidad (tórax acampanado y pectus) |
| Cuidados paliativos en la fase final de la enfermedad para aliviar disnea |
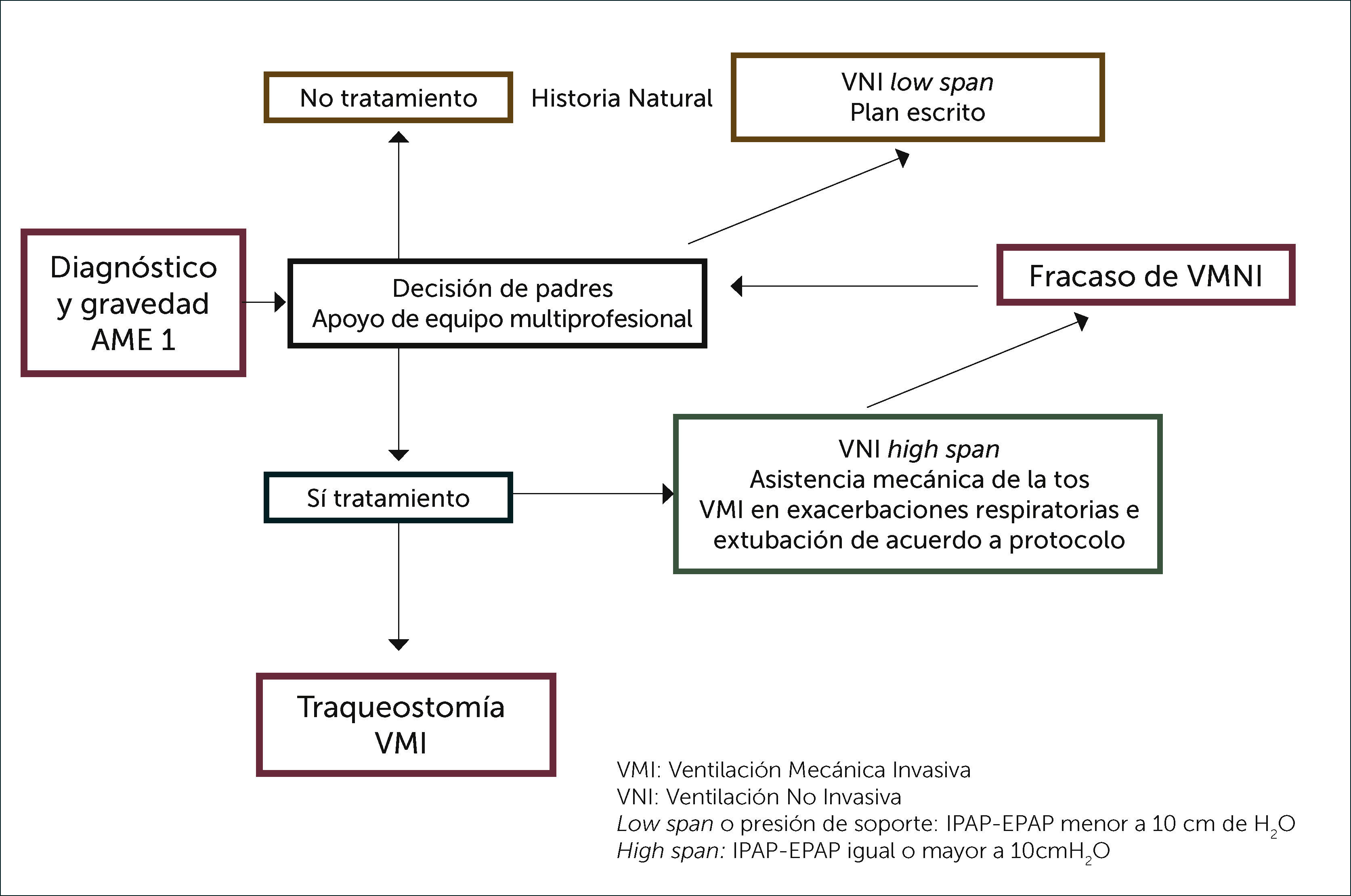
En la enfermedad de Werdnig Hofmann o AME 1 que es típicamente fatal antes de los 2 años, con un 50% de mortalidad a los 7 meses y 90% a los 12 meses, existe respiración paradojal, pectus y tórax acampanado si no son tratados en forma adecuada y oportuna. Las neumonías son frecuentes en relación a intercurrencias respiratorias virales y eventos aspirativos, debido a la tos inefectiva y al trastorno de la deglución 26,27. Las aproximaciones terapéuticas varían en los diferentes centros, siendo controversiales 28–31. Existen tres posibilidades en estos pacientes: seguir la historia natural con intervención mínima, traqueostomía y ventilación mecánica invasiva crónica o utilizar soporte ventilatorio no invasivo acompañado de la asistencia mecánica de la tos. La traqueostomía significará que no podrá respirar sin la ayuda del ventilador y no le permitirá desarrollar la habilidad fonatoria, a diferencia de la ventilación no invasiva, que si le permitirá la comunicación verbal, pero será muy dependiente de sus cuidadores, si bien es cierto las hospitalizaciones y sobrevida serán diferentes. Esta decisión debe ser tomada por los padres, acompañada por un equipo multidisciplinario que incluye el apoyo de un psicólogo conocedor de la enfermedad. Él tiene un rol facilitador en la toma de decisiones y ofrece contención emocional, acompañamiento a los padres y al equipo médico; debe corroborar que los padres del paciente y/o el niño entiendan la información entregada y la integren, para así tomar decisiones informadas, considerando su propio marco valórico.
De esta forma, podrán anticipar las consecuencias de sus decisiones e ir elaborando las dinámicas internas que irán surgiendo a lo largo de este proceso. Si la decisión es intermedia, como se ilustra en la Figura 3, una vez hecho el diagnóstico genético y ya comenzando la respiración paradojal, es necesario iniciar el apoyo ventilatorio no invasivo durante el sueño profundo con el fin de que el niño se aclimate.
.")
El ventilador portable debe ser siempre binivel, limitado por presión y/o por volumen 32–40. Las presiones inicialmente bajas, cercana a 8cmH20 aumentando rápidamente hasta lograr que desaparezca la respiración paradojal, alcanzando presiones sobre 14cmH20, manteniendo una span o presión de soporte -IPAP-EPAP- mayor a 10cmH20, con el fin de permitir el desarrollo adecuado de la caja torácica y pulmonar y facilitar la eliminación de CO2; el y EPAP cercano a 4 que ayuda a mantener la vía aérea alta abierta durante la fase respiratoria del ciclo, aumentando la CRF al reclutar alveolos, disminuir el trabajo respiratorio y permitir un gatillo más fácil de la VNI cuando existe un PEEP intrínseco o presión espiratoria positiva intrínseca, con un volumen corriente entre 8 y 12ml/kg. El ventilador debe tener frecuencia respiratoria de respaldo, entre 18 y 35 por minuto, dependiendo de la frecuencia respiratoria espontánea, de una o dos respiraciones bajo la espontánea en vigilia, que en la práctica se adiciona para prevenir la hipoventilación durante el sueño. Al poco tiempo los cuidadores reportan que los niños se acostumbran, hay menos diaforesis y despertares nocturnos. Esto puede realizarse a través de una interfase nasal. La ventilación debe ser continua durante las intercurrencias virales respiratorias y especialmente si la saturación de oxígeno está bajo 95%. Son pocas las complicaciones de la VNI: la disfunción bulbar y alteración de la deglución son contraindicaciones relativas y la falta de tolerancia a la interfase puede resolverse adecuando las máscaras nasales o almohadillas con materiales suaves para evitar las lesiones de la cara. Los padres han manifestado estar agradecidos de su uso. Es indispensable mantener una nutrición adecuada, y con frecuencia en los casos más severos aparecerá la alteración de deglución, para lo cual será necesario discutir las alternativas de alimentación futura ya sea por sonda nasogástrica, nasoyeyunal o planificar una gastrostomía en forma precoz, asociada o no a cirugía de Nissen. Se debe tratar el reflujo gastroesofágico y ocasionalmente es útil el glicopirolato para reducir las secreciones orofaringeas y salivación excesiva. El manejo domiciliario no invasivo es posible para estos pacientes.
Exacerbaciones respiratorias. Estas se asocian a infecciones respiratorias o aspiración; el desarrollo de obstrucción de la vía aérea por tapones mucosos y atelectasias por el aumento de secreciones favorece la hipoventilación alveolar e hipercapnia, por disminución del volumen corriente y el aumento de la frecuencia respiratoria.
Si la saturación de oxígeno baja de 95% podría deberse a la hipoventilación misma o a una alteración de la relación ventilación-perfusión por aumento de secreciones, atelectasias o neumonía. La fatiga muscular aumenta, favoreciendo aún más las atelectasias que llevan a hospitalización, intubación endotraqueal y, en ocasiones, la muerte.
Cuando aparece la respiración paradojal significa que ya es necesario el apoyo ventilatorio. En ocasiones la taquipnea y taquicardia pueden no ser evidentes. La infección respiratoria viral disminuye la capacidad vital y deja una debilidad que demora 3 a 4 semanas en recuperarse y a veces no regresa a su estado basal. Es por esto que los padres deben monitorizar la saturación de oxígeno y mantenerla sobre 95%, aumentando las maniobras de tos asistida y de VNI, con el fin de evitar hospitalizaciones. No se recomienda administrar oxígeno suplementario en la casa ya que esto puede aumentar, la hipercapnia. Si no logran revertir la saturación de oxígeno será necesario el manejo hospitalizado.
La administración de antibióticos en estos pacientes tiene un umbral más bajo que en niños sin esta patología. El manejo hospitalizado se debe realizar en los pacientes que no logran saturaciones sobre 94% con aire ambiental, luego de asistencia de la tos intensiva, o si no cuentan con estos recursos. Sería necesario descartar mediante una radiografía de tórax la existencia de atelectasia o neumonía, tomar exámenes en busca de agentes etiológicos y gases arteriales. En aquellos usuarios de VNI nocturna será necesario aumentarla a las horas diurnas, con el fin de evitar la intubación endotraqueal y la ventilación mecánica invasiva. Si la intubación es inevitable, podría extubarse hacia la VNI continua con un span sobre 10cmH20 cuando la saturación de oxígeno sea >94% en aire ambiental y la pCO2 sea normal, las secreciones sean escasas, no exista atelectasia, siguiendo el protocolo de Bach 33,35. Si el paciente aún requiere oxigenoterapia para mantener saturación arterial sobre 95% debe intensificarse la kinesiterapia respiratoria y el uso del asistente de la tos. El paciente se intentará extubar a su interfase habitual, pero en ocasiones sería necesario utilizar transitoriamente una interfase facial. Después de la extubación se debe mantener la nutrición intensiva, kinesiterapia, uso del asistente de la tos frecuente y aspiración de secreciones nasofaríngeas (Tabla 4). Se debe evitar la oxigenoterapia adicional a menos que se administre, si es necesario, durante las sesiones de kinesiterapia 40.
PROTOCOLO DE EXTUBACIÓN EN ATROFIA MUSCULAR ESPINAL
| Administración de oxígeno limitada hasta lograr saturación de oxígeno de 95% |
| Uso de asistente mecánico de la tos o In-Exsufflator (MI-E) a través del tubo endotraqueal tan frecuente como sea necesario para revertir la desaturación |
| Aspiración endotraqueal y por boca posterior a su uso según necesidad |
| Extubación si cumple los siguientes criterios: afebril, sin signos de infección bacteriana, sin requerimientos de oxígeno, mejoría de la radiografía de tórax, sin depresores respiratorios, mínima necesidad de aspiración, idealmente menos de 1 a 2 cada 8 horas, escasa coriza. |
| Extubación hacia ventilación no invasiva sin requerimientos de oxígeno |
| Monitorización de la saturación de oxígeno como guía de drenaje postural, kinesiterapia respiratoria y uso de asistente mecánico de la tos para revertir cualquier desaturación debido a un tapón mucoso |
| Si hay retención de CO2 o dificultad en la sincronización con el ventilador, se debe aumentar la presión-volumen del ventilador sin aumentar la frecuencia respiratoria. Si se mantiene la desaturaciòn implica necesidad de reintubar |
| Luego de reintubar, el protocolo se utiliza para un segundo intento de extubación |
| Alta una vez que la saturación esté estable por 24-48 horas |
Adaptado de Bach J. The use of mechanical ventilation is appropriate in children with genetically proven spinal muscular atrophy type 1: the motion for. Paediatric Respiratory Reviews 2008;9,45-50.
La sobrevida de pacientes con AME ha mejorado gracias a los cuidados respiratorios descritos, ya sea con traqueostomía y VMI, VNI, uso de asistente mecánico de la tos y de alimentación suplementaria. Oskoui publicó que la sobrevida de niños con AME 1 aumentó entre aquellos nacidos entre el período 1980–1994 y 1995–2006 desde una mediana de 7.5 meses hasta 24 meses y una probabilidad de sobrevida a los 24 meses de 31 a 74% 7. Gregoretti reportó la sobrevida de 194 pacientes con AME 1 cuyos padres eligieron entre las diferentes alternativas de cuidados respiratorios: 62.3% prefirió la evolución natural de la enfermedad, 21.7% manejo ventilatorio invasivo a través de traqueostomía y 16% en manejo no invasivo que incluyó VNI y asistencia mecánica de la tos. La elección de traqueostomía disminuyó a un 12% entre los años 2005 y 2010. La sobrevida aumentó en aquellos con asistencia ventilatoria a 67 y 89% a los 2 años y de 45 y 90% a los 4 años para el manejo no invasivo e invasivo respectivamente 42.
DESARROLLO DE NUEVAS TERAPIASEl objetivo de todas las nuevas terapias es aumentar los niveles de la proteína SMN por medio de distintas estrategias:
- •
Reemplazar o corregir el gen SMN1 defectuoso
- •
Modular los reguladores del splicing para promover la inclusión del exón 7 en el gen SMN2 y activar los promotores del gen SMN2
- •
Estabilizar la proteína SMN
Otras estrategias terapéuticas incluyen terapia con células madres, moléculas neuroprotectoras y compuestos que aumentan la fuerza muscular. El desarrollo de intervenciones terapéuticas futuras dependerá de una comprensión más acabada de otros factores modificadores del fenotipo de la enfermedad, aparte del ya conocido gen SMN2. Los estudios preclínicos, realizados en el modelo de ratón “delta 7” portador del transgen SMN2 humano, nos ha enseñado que mientras más precoz es el reemplazo de la proteína SMN normalmente disminuida en este cuadro, los efectos en el grado de reversión del fenotipo son más importantes. Esto sugirió la existencia de ventanas terapéuticas estrechas que han debido ser consideradas en los diseños y criterios de inclusión de los ensayos clínicos actualmente en curso. Los potenciales efectos del aumento de la proteína SMN en etapas más tardías, en fenotipos clínicos menos severos de la enfermedad, donde cambios menores en la fuerza muscular pueden modificar en forma significativa funciones que mejoran la vida de los pacientes, se encuentra aún en etapa de estudio. Hoy existen más de 17 ensayos clínicos en AME en diferentes etapas preclínicas y clínicas.
Terapia génica. En los últimos años varios grupos de investigación han demostrado con éxito la transfección del gen SMN1 a través de la inyección endovenosa sistémica de un virus adeno-asociado (scAAV). El reemplazo de gen SMN1 por un vector viral transportador del gen humano será con mucha probabilidad, exitoso en restaurar los niveles de proteína SMN. Este vector logra traspasar la barrera hematoencefálica e infecta eficazmente las motoneuronas logrando de esta manera restaurar niveles más adecuados de proteína SMN, con la ventaja de que este vector viral no se integra al genoma del huésped. Después de pasar con éxito los ensayos preclínicos el año 2014, se dio inicio al primer ensayo clínico en humanos para la administración sistémica de AAV9, habiéndose completado la fase 1 en nueve lactantes AME tipo 1 menores de 9 meses de edad de que recibieron una dosis única de este vector viral. En agosto de 2016 la compañía aveXis 43 informó que 11 de 12 pacientes (92%) y 8 de 12 pacientes (67%) en la cohorte 2 que es la que recibió mayor dosis del vector viral, habían logrado puntuaciones de al menos 40 o 50 puntos, en la escala CHOPINTEND respectivamente y 3 de 12 pacientes (25%) de esta misma cohorte 2 lograron una puntuación de 60, lo que es considerada rango normal.
Oligonucleótidos antisentido. Los oligonucleótidos antisentido (ASO's) son moléculas creadas con el objetivo de promover la inclusión del exón 7 en el transcrito del gen SMN2. Dos de estas moléculas, morfolino y 2¿-O-metoxietil (MOE) pueden bloquear el silenciador intrónico del splicing N1 (ISS-N1) de SMN2 permitiendo la inclusión del exón 7 en el transcrito de SMN2. Los estudios preclínicos con estas moléculas antisentido han mostrado una mejoría substancial de la sobrevida del fenotipo clínico de AME en modelos de ratón Smn*7 desde un promedio de 13 días hasta mas de 100 días con una dosis única de administración 44. Los resultados de los ensayos clínicos en fase I-II de Isis Pharmaceuticals con su molécula antisentido MOE (ISIS-SMNRx) demostró ser eficaz. Esta compañía está trabajando actualmente en 2 ensayos clínicos fase III con ISIS-SMNRx: uno es ENDEAR, que evalúa la eficacia y seguridad clínica de la administración intratecal de la molécula y otro es CHERISH, ensayo clínico multicéntrico doble ciego randomizado en 117 niños con AME no ambulantes. Uno de los oligonucleótidos antisentido, nusinersen (ASOs) del laboratorio ISIS pharmaceutical ha publicado ya los resultados de su estudio fase 1 9. Esta molécula se une al pre-mRNA de SMN2 y promueve la inclusión del exón 7 en el transcrito aumentando la producción y función de la proteína SMN. Esta molécula se administra por vía intratecal y es el primer ensayo clínico abierto de dosis única creciente aprobado en niños con AME.
CONCLUSIONESLa complicación más seria en AME es la insuficiencia respiratoria cuyo enfoque debiera ser preventivo con el manejo intensivo de la tos, evitando la deformación de la caja torácica y permitiendo el desarrollo pulmonar, tratando la hipoventilación, manejando oportunamente las infecciones respiratorias, el trastorno de deglución, el reflujo gastroesofágico y la malnutrición. Los avances en el conocimiento de la patogénesis y la historia natural de la AME, sumadas a nuevas estrategias terapéuticas y al manejo en equipo multidisciplinario de acuerdo a los estándares de tratamiento, permite mejorar la calidad y expectativa de vida de estos pacientes, evitando situaciones difíciles para las familias, a la espera de las terapias curativas.
Al equipo de trastornos motores CLC por sus comentarios.
Las autoras declaran no tener conflictos de interés, en relación a este artículo.