



En enfermedad renal crónica (ERC) el riesgo aumentado de muerte por causa cardiovascular se relaciona con calcificaciones vasculares precoces y progresivas. Estas calcificaciones ocurren por un proceso altamente regulado. En los vasos de los pacientes con ERC coexisten calcificaciones en la íntima vascular asociadas a ateroesclerosis, pero además hay calcificaciones en la túnica media del vaso. Estos dos tipos de calcificaciones tienen distintas consecuencias fisiopatológicas cardiovasculares. Los principales componentes que interactúan para producir calcificación de la matriz extracelular son la célula muscular lisa vascular (CMLV), factores que inician el proceso de transformación osteocondrogénica, inductores e inhibidores de la calcificación, factores que regulan a estos últimos. Como respuesta al proceso de calcificación, el vaso expresa inhibidores que disminuyen la diferenciación osteoblástica, amplificando la lesión ósea adinámica. Así, el conocimiento del proceso patológico bidireccional del eje Hueso-Vaso ayuda a explicar la estrecha correlación entre pérdida de masa ósea y calcificación vascular.
The elevated cardiovascular mortality risk in chronic renal disease (CKD) patients is associated to accelerated vascular calcifications. This is an actively regulated process. Intimai calcifications associated to atherosclerosis, and medial vascular calcifications are found in CKD patients. Each of these two types of calcifications is associated to different cardiovascular physiopathological consequences. Main components involved in vascular calcification are vascular smooth muscle cells (VSMCs), factors involved in the osteochondrogenic transdifferentiation, inducers and inhibitors of vascular calcification, and their regulators. In response to calcification, vessels produce inhibitors that will decrease bone formation and could be involved in the pathogenesis of adynamic bone disease. It is suggested that the Bone-Vascular cross-talk is bidirectional, and as well it could be involved in the strong correlation between bone loss and vascular calcification.
La enfermedad renal crónica (ERC) constituye un problema de salud pública mundial. Existe un incremento en la prevalencia e incidencia de ERC terminal. La magnitud del problema aumentará en los próximos años en relación con el envejecimiento de la población y la elevada prevalencia de diabetes e hipertensión arterial. En Chile ha alcanzado una incidencia de alrededor de 1.000 pacientes nuevos por año que ingresan a un programa de diálisis crónica y una prevalencia actual de aproximadamente 17.000 enfermos en este programa. Mientras la cobertura de un programa de sustitución renal a los enfermos con ERC terminal en nuestro país ha mejorado notablemente entre 1980 y 2012 desde 12.7 a 1,001 pacientes/millón de habitantes, la mortalidad se mantiene elevada, tal como sucede en países desarrollados, con tasas aproximadas de 15% anual.
La principal causa de muerte en pacientes con ERC y ERC en diálisis es eventos cardiovasculares (CV), con una mortalidad 30 a 40 veces mayor a la observada en la población general en sujetos menores de 40 años (1). Los enfermos con ERC tienen una alta probabilidad de morir por eventos CV antes de ingresar a una terapia de sustitución renal (2). Las causas bien establecidas de riesgo cardiovascular observadas en la población general no explican del todo el riesgo aumentado en la ERC. En general, los mecanismos asociados a eventos CV en ERC son hipertensión arterial, dislipidemia, activación del sistema renina-angiotensina (SRA), disfunción endotelial, stress oxidativo, inflamación crónica y calcificaciones vasculares. Concomitantemente, alteraciones propias del estado urémico contribuyen a este aumento de riesgo CV, como sobrecarga de volumen, anemia y miocardiopatía. De los pacientes que inician diálisis, un 40% tiene evidencia de enfermedad coronaria y 85% tiene ya una estructura y función anormal de su ventrículo izquierdo.
El estudio de Framingham fue el primero en establecer la asociación entre ERC y eventos/muerte CV en la población general. Go y cols. evaluaron la relación entre tasa de filtración glomerular (FG) y eventos adversos CV en una población de bajo riesgo (3). Después de ajustar por edad, sexo, raza, comorbilidades y estado socioeconómico, se observó un incremento en cada uno de los 3 outcomes primarios (muerte por cualquier causa, eventos CV y hospitalizaciones) para cada etapa de FG disminuida. Lamentablemente, en la mayoría de los estudios epidemiológicos de riesgo CV en la población, los pacientes con ERC son excluidos en el diseño original y en los pocos pacientes con ERC que finalmente participaron se hace posteriormente análisis post hoc. Un ejemplo de ello son los estudios HOT y HOPE. El estudio HOT (Hypertension Optimal Treatment) mostró que el riesgo relativo ajustado para eventos CV mayores fue de 1.65 para sujetos con FG <60ml/min, comparado con 1.58 para aquellos con FG >60ml/min (4). El estudio HOPE (Heart Outcomes and Prevention Evaluation) reveló que los 980 sujetos con ERC leve tuvieron una incidencia acumulativa de eventos CV de 22.2% vs 15% en aquellos con función renal normal (5).
Enfermedad cardiovascular en pacientes con enfermedad renal crónicaDesde los inicios de los programas de hemodiálisis crónica, se observó que estos pacientes desarrollan enfermedad vascular ateroesclerótica más precoz y frecuentemente que la población general. A los factores de riesgo clásicos, tales como hipertensión, diabetes, dislipidemia, obesidad, tabaquismo y antecedentes familiares, se deben agregar otros factores que son particularmente importantes en la ERC: productos finales avanzados de la glicación (AGE), estrés oxidativo, óxido nítrico (NO), dimetilarginina asimétrica (ADMA), homocisteína, anemia, fósforo y producto calcio-fósforo (6). Todos ellos participantes en la disfunción endotelial, que es reconocida como uno de los mecanismos iniciales de la ateroesclerosis en estos enfermos. Hay evidencia experimental que sugiere que la disfunción endotelial microvascular que participa en los mecanismos que llevan a ERC, es también responsable de acelerar la aterogénesis.
Durante los últimos años se ha puntualizado que esta elevada tasa de riesgo CV se asocia estrechamente a la presencia de calcificaciones vasculares en esta población (7). Las calcificaciones vasculares se observan en población normal y han sido consideradas como una condición propia del envejecimiento. En la población con ERC las calcificaciones vasculares ocurren en forma precoz y progresan rápidamente en concordancia con su elevada tasa de enfermedad CV prematura. Estudios de auptosias en 120 niños urémicos revelaron la presencia de calcificación en tejidos blandos en más del 60% de los casos y de ellos el 60% tenía calcificación vascular. Goodman y cols. estudiaron la presencia de calcificación coronaria mediante técnica de EBCT (electron bean computed tomography) en menores de 30 años en diálisis crónica y encontraron que el 80% de ellos tenía una calcificación coronaria severa y progresiva. La magnitud de la calcificación coronaria se correlacionó con hi-perfosfatemia, aumento del producto calcio χ fósforo sérico y elevada ingesta de calcio diario, dependiente este último del consumo de quelantes de fósforo basados en sales de calcio (8).También se han descrito calcificaciones extraóseas bajo condiciones que no son explicadas exclusivamente por alteraciones del metabolismo calcio-fósforo.
La existencia de calcificaciones aórticas es un predictor de morbimor-talidad cardiovascular independiente de los factores de riesgo clásicos. Recientemente, en un estudio en 6.722 sujetos sin ERC (22% hispánicos) se determinó que la calcificación coronaria, medida por tomografía computada, fue un predictor importante de enfermedad coronaria. Diversos estudios han demostrado que el tejido cardiovascular de los sujetos en diálisis crónica está marcadamente calcificado. Rostand y cols. mostraron que el contenido de calcio del miocardio de pacientes en diálisis crónica fue significativamente más elevado que el de sujetos no urémicos (9). Del mismo modo, se ha demostrado una mayor prevalencia de calcificaciones en válvula mitral y aórtica en esta población, lo que se asocia a una mayor mortalidad.
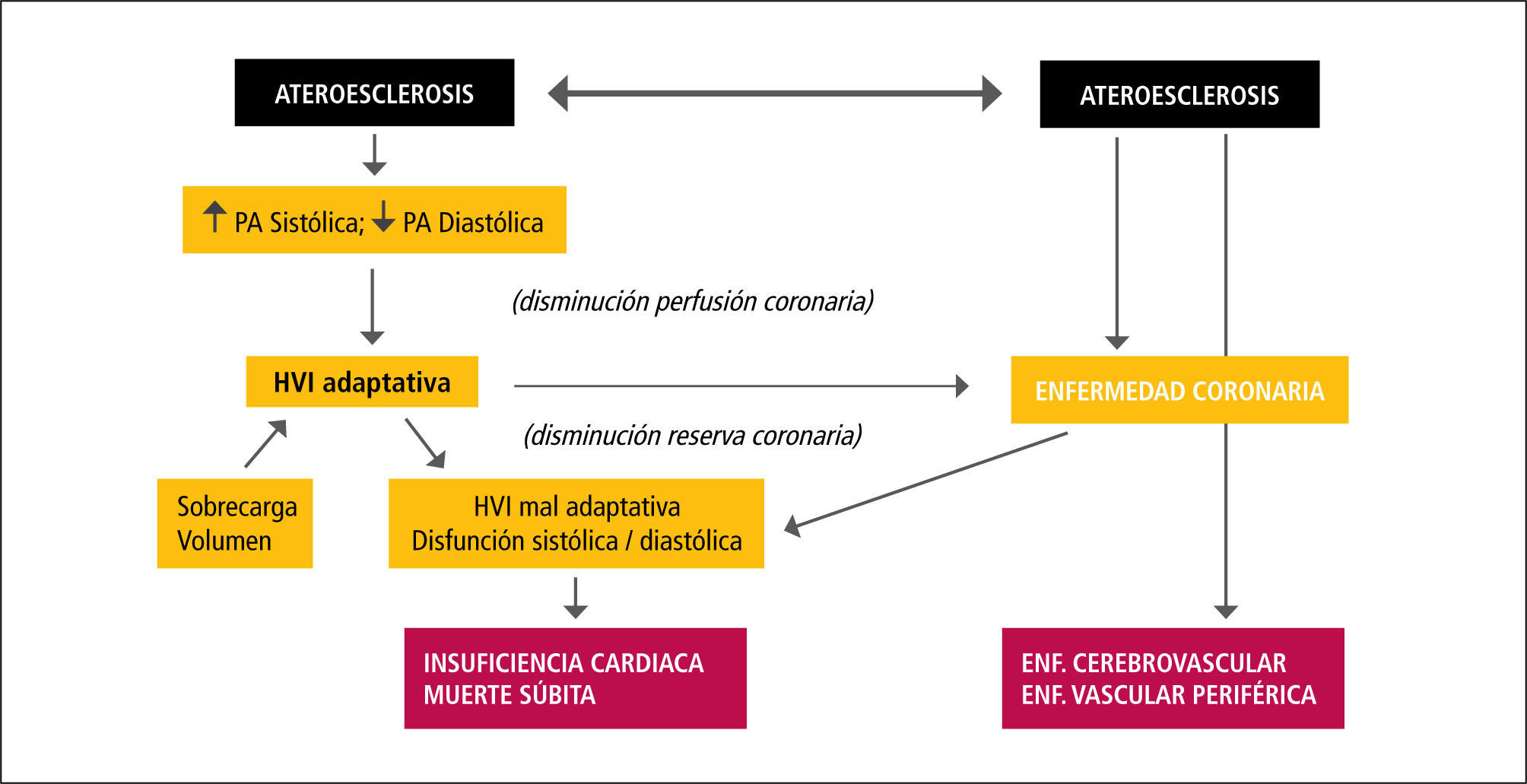
Inicialmente se estimó que el proceso de calcificación vascular observado en la ERC era producto de un depósito pasivo de calcio y fósforo derivado de una severa alteración metabólica persistente en el tiempo y que se amplifica por la mayor sobrevida de los enfermos urémicos al iniciarse diálisis. Este depósito en los vasos ocurriría en la región luminal, mayormente asociado a la placa ateroesclerótica. Mientras la calcificación de la intima del vaso está asociada a la placa ateroesclerótica, en la gran mayoría de los enfermos urémicos la calcificación se observa inicialmente en la túnica media del vaso (calcificación medial o de Mönckeberg), generalmente en la lámina elástica interna y no se asocia a macrógafos cargados de lípidos o hiperplasia intimai y afecta principalmente a las arterias elásticas (10-12). Esta calcificación ocurre en la forma de hidroxiapatita y se produce en la matriz entre las células y no dentro de las células, a diferencia de la denominada calcificación distrófica. Ambos tipos de calcificación son muy comunes en la ERC avanzada y pueden coexistir en un mismo vaso. Como consecuencia de ello se ven afectadas las dos funciones arteriales interrelacionadas: aportar un flujo sanguíneo adecuado a los tejidos y órganos (función de conducción), y transformar las oscilaciones cíclicas de alto flujo y presión en la aorta en un flujo capilar continuo y de baja presión (función de amortiguación). La ateroesclerosis, caracterizada por placas ateromatosas que restringen el flujo sanguíneo y cuya consecuencia es la isquemia o infarto de los tejidos perfundidos, es la principal causa de trastorno de la capacidad conductiva del vaso y una causa frecuente de enfermedad isquémica miocárdica, accidentes vasculares cerebrales y enfermedad vascular periférica. Arterioesclerosis es responsable del compromiso de las propiedades elásticas del vaso (trastorno de la amortiguación) derivado de la calcificación de la túnica media. Este trastorno determina una reducción en la distensibilidad aórtica, aumento de la postcarga miocárdica e hipertrofia ventricular izquierda, alteraciones que confieren un alto riesgo de mortalidad (Figura 1). La rigidez arterial es considerada actualmente un potente predictor independiente de mortalidad cardiovascular en pacientes con ERC terminal en hemodiálisis (13,14).

La presencia de calcificaciones extra óseas se asocia estrechamente a los trastornos del metabolismo mineral y óseo que se observa en el paciente con ERC. Las calcificaciones arteriales no sólo se producen por una reabsorción ósea aumentada, debido a niveles elevados de hormona paratiroidea, sino también por una baja actividad ósea y una enfermedad ósea adinámica asociada.
Patología de la calcificación vascularDurante los últimos años se ha reconocido que la calcificación vascular es un proceso activo y regulado, en el que intervienen diferentes mecanismos no excluyentes entre sí. Los niveles elevados de Ca, R y CaxP promueven la formación y crecimiento de núcleos de cristales de hidroxiapatita. La hidroxiapatita es el principal componente mineral de los huesos. El origen de las células responsables de la mineralización en la pared vascular es aún desconocido, aunque las evidencias experimentales señalan a la célula muscular lisa vascular (CMLV) y no a células pasajeras, como la célula de estirpe osteoblástica que participa en el proceso de mineralización. Osteoblastos y CMLV son células diferenciadas a partir de una misma célula mesenquimal pluripotencial. Ratones knockout para Core binding factor alpha 1 (Cbfa-1) no forman hueso mineralizado, lo que indicaría que Cbfa-1 es indispensable para que la célula troncal mesenquimal se transforme en osteoblasto (15). Cbfal es el principal regulador de la diferenciación ósea. Actúa como un factor de transcripción que dispara la expresión de importantes genes de la línea osteoblástica como son la osteocalcina, la osteopontina, la fosfatasa alcalina o el colágeno tipo I. El fosfato y las toxinas urémicas regulan aumentando su expresión.
En el hueso las células madres mesenquimales se diferencian a osteoblastos bajo la acción de factores de diferenciación como el BMP-2, que también se expresa en la pared de la arteria calcificada.
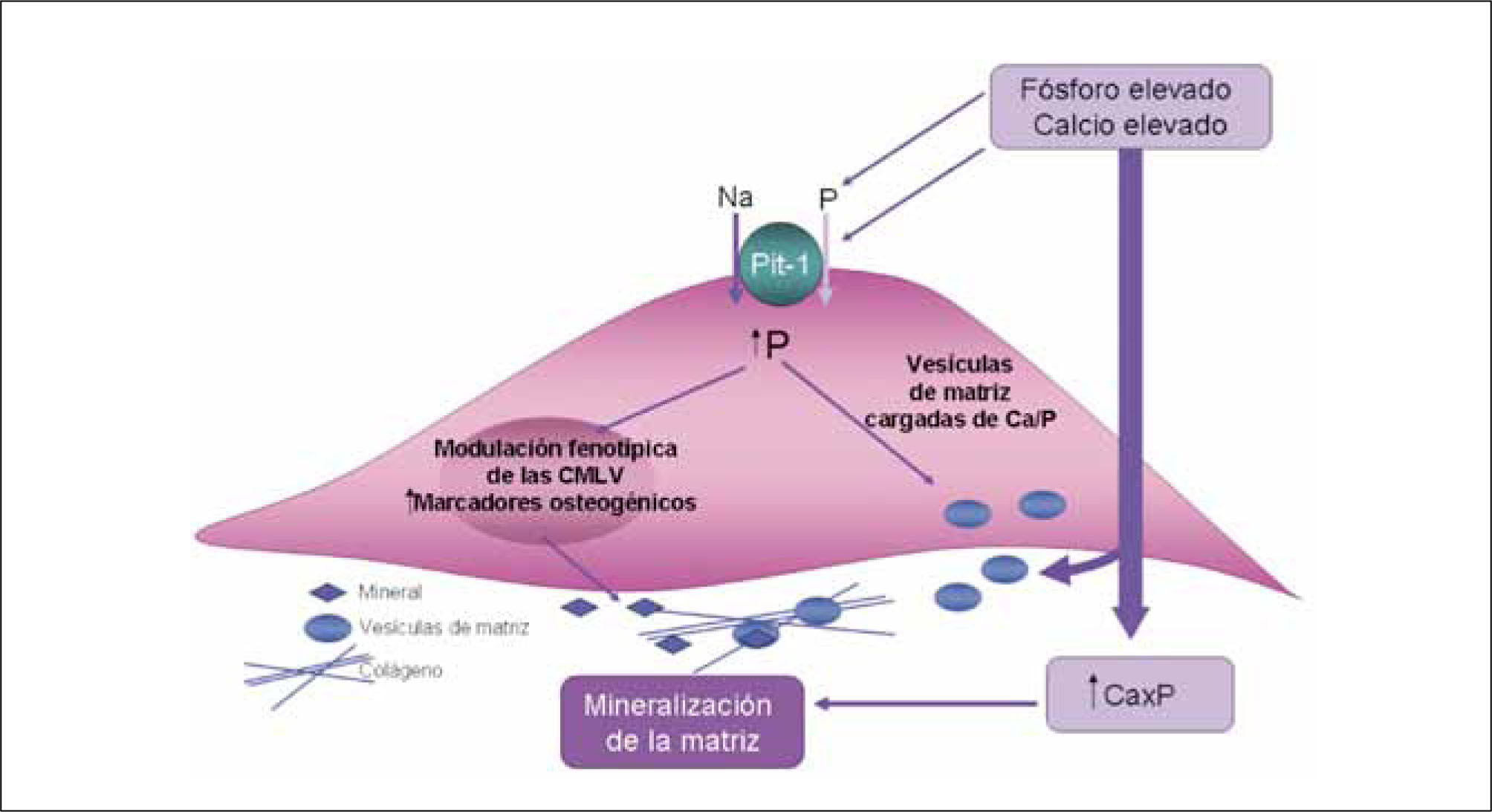
En estudios in vitro se ha observado que al exponer las CMLV a altas concentraciones de fósforo o calcio se produce el depósito de hidroxiapatita en la matriz extracelular (16). Aún más, cuando se incuba con ambos elementos a la vez, se observa un efecto sinérgico de la calcificación. Este fenómeno no es simplemente un proceso pasivo de precipitación de Ca y P, sino que implica un cambio fenotípico de las CMLV y la regulación de genes comúnmente asociados a la diferenciación ósea. El cotransportador de fosfato dependiente de sodio, NaPi, cuyo subtipo Pit-1 se encuentra presente en las CMLV, es un factor importante en la mediación de esta diferenciación osteocondrogénica. Niveles altos de fósforo estimula la actividad del cotransportador y la carga, mientras niveles elevados de calcio induce la expresión de mRNA de Pit-1. El aumento de la expresión de genes osteogénicos provoca la secreción de moléculas minerales (vesículas de matriz, proteínas ligadoras de calcio, fosfatasa alcalina y matriz extracelular rica en colágeno) (17) (Figura 2). La mayoría de los investigadores están de acuerdo en el rol esencial de la CMLV en el proceso de la calcificación celular y hay muchos mecanismos que cooperan y amplifican esta respuesta de transformación osteocondrogénica, elastinosis y calcificación de matriz extracelular:

Modelo de los efectos del calcio y el fósforo sobre la mineralización de las CMLV
La calcificación vascular está relacionada con la aparición de pequeñas vesículas de matriz con contenido citoplásmico y membrana celular intacta (al igual que sucede en el desarrollo óseo); estas vesículas se forman a partir de células donde se origina mineralización o son el resultado del proceso de apoptosis celular (cuerpos apoptóticos). La pared de los vasos del paciente urémico está lesionada por procesos de inflamación y estrés oxidativo, por lo tanto es razonable pensar que exista apoptosis celular. El laboratorio de C. Shanahan ha mostrado que la apoptosis de CMLV regula la calcificación vascular in vitro e in vivo (17). De acuerdo a estos autores, las vesículas de matriz son capaces de concentrar calcio en su interior y son el origen de los cristales de hidroxiapatita.
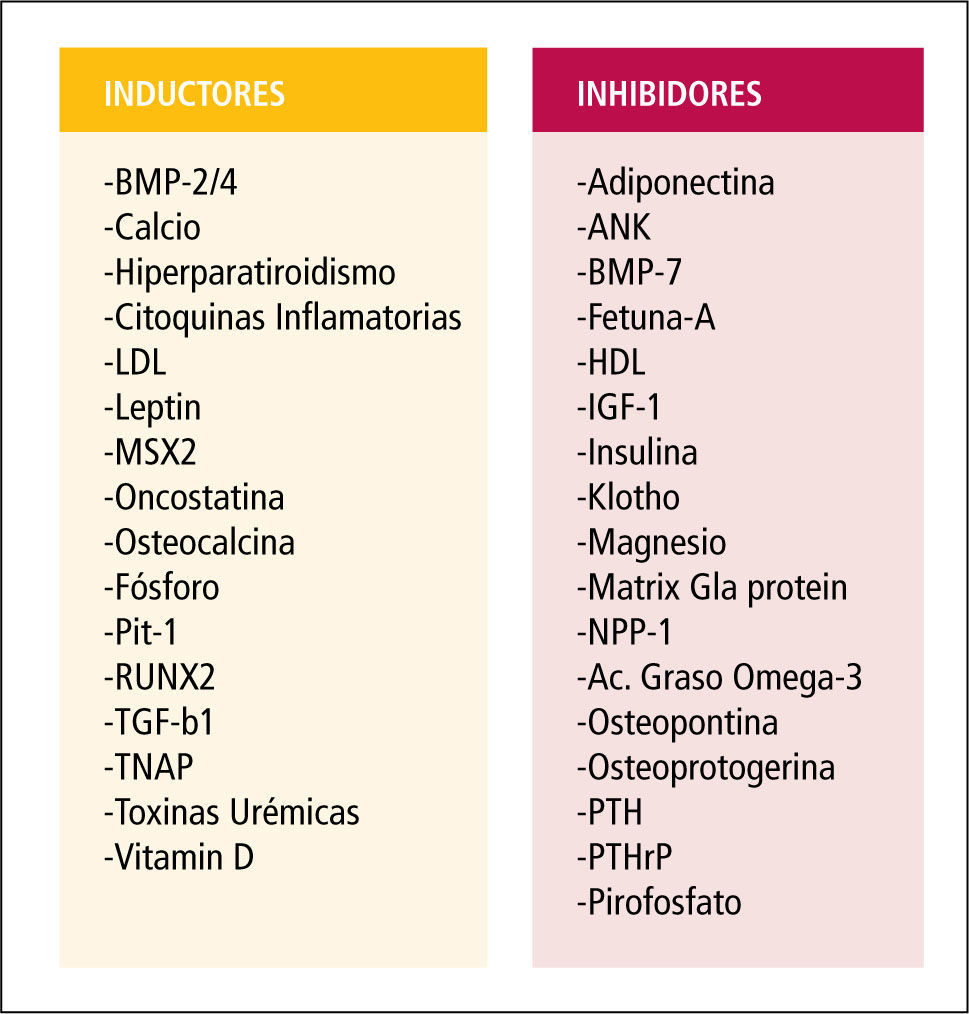
Inhibidores de la calcificaciónLas células de los vasos expresan en condiciones normales moléculas inhibidoras de la mineralización (Figura 3). La pérdida de su expresión, como sucede en la ERC, provoca una calcificación espontánea y un aumento de la mortalidad. Mediante análisis de mutaciones en ratones se ha elaborado una lista con estas moléculas inhibidoras de la calcificación (11), entre las que se incluye a:

Lista de inductores e inhibidores de la calcificación vascular
- Matrix Gla Protein (MGP). La MGP fue el primer inhibidor de la calcificación identificado. Es una proteína dependiente de vitamina K que se expresa constitutivamente en CMLV, y también en células endoteliales, de vasos normales pero está disminuida en arterias calcificadas. Se ha observado que su expresión disminuye en modelos in vitro de calcificación. La administración de warfarina a ratas produjo una calcificación de arterias similar a la observada en el ratón deficiente para MGP, lo que sugiere que warfarina induce calcificación arterial por inhibir la γ-carboxilación de MGP. Los niveles séricos de MGP son menores en pacientes con calcificaciones que en los que no tienen. Además, los ratones knockout para MGP desarrollan severas calcificaciones de la media y mueren por ruptura aórtica. Se considera a MGP como el principal inhibidor tisular de calcificación. Una de las funciones principales de la proteína GLA es desactivar BMP-2 y evitar que actúe en la pared del vaso como factor diferenciador de células de estirpe osteoblástica. Los pacientes con ERC que reciben tratamiento anticoagulante con dicumarínicos se encuentran en mayor riesgo de desarrollar calcificación vascular. Actualmente están en proceso estudios clínicos con diseños estadísticos robustos para determinar si la administración de vitamina K a pacientes con ERC disminuye el riesgo de calcificación vascular.
- Fetuina-A. Es una glicoproteína sérica que inhibe la calcificación vascular ectópica. Es un potente inhibidor de la formación de hidroxiapatita, reduciendo la formación de cristales en soluciones que contienen calcio y fósforo in vitro. Inhibe la formación de estos cristales pero no afecta a los ya formados. Los ratones deficientes en esta proteína desarrollan extensas calcificaciones de los tejidos blandos como miocardio, riñón, lengua y piel. Se le considera el principal factor inhibidor circulante de calcificación vascular. La inhibición de fosfato cálcico básico es efectuada por la formación transitoria de esferas coloidales que contiene fetuina, calcio y fosfato, denominadas calciproteínas, y es el principal componente de una masa compleja de alto peso molecular que contiene Ca, R y MGP. La fetuina-A se sintetiza en el hígado y constituye la mayor parte de la banda α-2 de la electroforesis de proteínas. Fetuina-A es regulada como un reactante negativo de fase aguda, por lo que en procesos inflamatorios se encuentra disminuida. Los niveles séricos de fetuina-A están disminuidos en pacientes en hemodiálisis crónica y los niveles bajos se correlacionan con elevación de los niveles de proteína C reactiva y con mortalidad cardiovascular.
- Osteopontina (OPN). La OPN es una fosfoproteína que se encuentra normalmente en los tejidos mineralizados como hueso y dientes, y participa en la regulación de la mineralización al actuar como inhibidor del crecimiento de los cristales de hidroxiapatita. Aunque no se encuentra en arterias normales, algunos autores han detectado su expresión en placas ateroscleróticas y válvulas aórticas calcificadas. Osteopontina también se expresa en abundancia en las zonas vasculares con calcificación e inhibe la cristalización de hidroxiapatita in vitro y la calcificación en CMLV en cultivo. Ratones deficientes en osteopontina no desarrollan calcificación vascular pero la ausencia de OPN acelera la calcificación en ratones deficientes de MGP. Estos estudios señalarían que aunque OPN no es un inhibidor endógeno de calcificación en vasos normales, es abundante en vasos calcificados y modula la calcificación en estados más avanzados. Giachelli y cols cruzaron ratones OPN-/- (que no muestran manifestaciones vasculares) con ratones MGP-/- (que si desarrollan calcificaciones vasculares) y observaron que los ratones doble knockout OPN-/-MGP-/- mostraron una calcificación más acelerada que el grupo sólo knockout para MGP. Estos estudios indicarían que OPN es un inhibidor inducible de la calcificación vascular in vivo.
- Osteoprotegerina (OPG). Es un miembro de la familia de receptores de los factores de necrosis tumoral (TNFR), que ha sido identificado como regulador de la reabsorción ósea. OPG es producido por una gran cantidad de tejidos, incluidos el sistema cardiovascular, pulmón, riñón, sistema inmune y el hueso. En las lesiones calcificadas avanzadas OPG se presenta alrededor del área calcificada. Se ha observado que los ratones deficientes en OPG desarrollan osteoporosis severa y calcificación de la media vascular, lo que sugiere un rol como inhibidor de la calcificación vascular. OPG regula la activación osteoclástica y su déficit en ratones lleva a calcificación vascular. En pacientes con ERC terminal los niveles plasmáticos de OPG son elevados y correlacionan con el grado de calcificación vascular.
OPG funciona como un receptor soluble, señuelo de ligando (RANKL) del receptor activador del factor nuclear – kB (RANK). RANKL es producido por el osteocito y osteoblasto y estimula RANK en células precursoras de osteoclasto, fundamental entonces en el proceso de orquestar el remodelamiento óseo. RANKL también es producido por las células T activadas y esto permite, entre otras, aumentando los mediadores de inflamación. OPG es además receptor del ligando inductor de la apoptosis relacionado con el factor de necrosis tumoral (TRAIL), que es un potente inductor de apoptosis. TRAIL se encuentra en gran variedad de tejidos, incluyendo la CMLV y células endoteliales. En las lesiones ateroescleróticas humanas TRAIL se ha localizado en torno a las áreas calcificadas.
- Pirofosfato. La concentración elevada de pirofosfato (PPi) previene la formación de cristales de hidroxiapatita y calcificación. Ratones que carecen de la enzima pirofofatasa fosfo-diesterasa-1 desarrollan un fenotipo alterado de la CMLV y calcificación vascular. En ERC terminal puede ocurrir un déficit por remoción de PPi durante hemodiálisis (19). Los bifosfonatos son análogos de pirofosfatos, que a diferencia de éste -de muy corta vida media- tiene mayor periodo de acción. Hay fuerte evidencia experimental y hay casos clínicos aislados que indican que bifosfonato inhibe la calcificación vascular en animales y humanos. Su administración en pacientes con ERC debe considerar que el uso de bifosfonatos tiene el riesgo de afectar el remodelamiento óseo y producir enfermedad ósea adinámica, lo que no sólo dañará al hueso sino que amplificará el proceso de calcificación vascular.
Activadores de la calcificaciónEn el suero de los pacientes con ERC existen sustancias capaces de estimular la calcificación, además de hiperfosfemia e hipercalcemia. CMLV bovinas en presencia de suero urémico incrementan la expresión de proteínas relacionadas con el proceso de calcificación. Se ha identificado un amplio número de factores de la uremia que son capaces de inducir genes osteogénicos, de transformación osteoblástica y de secreción de algunas proteínas de la matriz ósea en la pared de los vasos y tejidos blandos. Algunos de estos factores son: el factor de necrosis tumoral (TNF), citoquinas inflamatorias, fibronectina, colágeno tipo 1 y 25-hidrocolesterol (18) (Figura 3). Estas sustancias del suero urémico promueven la expresión de moléculas fundamentales para la calcificación vascular como Fosfatasa alcalina (ALP). Es uno de los marcadores fenotípicos de los osteoblastos y se le considera esencial en el proceso de calcificación vascular. Se ha detectado su presencia en las calcificaciones vasculares de íntima, media, y de válvulas cardiacas. ALP expresada en la superficie celular puede actuar sobre los liberadores de fosfato, liberando fosfato inorgánico y también degrada pirofosfato. Las citoquinas inflamatorias y la vitamina D inducen su aumento y la mineralización.
Rol del fósforo en la calcificación vascularLa hiperfosfemia es un trastorno metabólico frecuente e intenso en los enfermos con ERC y es uno de los principales factores patogénicos del hiperparatiroidismo secundario (HPT 2o). La hiperfosfemia, independiente de HPT 2o, ha sido ligada a un riesgo aumentado de mortalidad cardiovascular en pacientes en diálisis. Ambos, hiperfosfemia e HPT 2o, han sido estrechamente correlacionados en forma independiente con la calcificación de la pared vascular en enfermos urémicos. La elevación del producto calcio-fósforo se ve potenciada por el uso de medidas para controlar tanto hiperfosfemia como HPT 2o, con quelantes de fósforo a base de sales de calcio y 1,25-(OH)2-vitamina D3 respectivamente (20, 21). Ha sido demostrado en estudios in vitro, usando cultivos primarios de CMLV, que el aumento de la concentración de fósforo extracelular induce a la célula muscular lisa a producir calcificaciones con transformación del fenotipo vascular de estas células a un fenotipo osteogénico, con la expresión de osteo-calcina y Cbfa-1. Estos efectos se observaron con niveles de fósforo extracelular muy similares a los de pacientes en diálisis (2,0mM/L). De este modo, la inducción de marcadores de células de estirpe osteoblástica como osteocalcina y Cbfa-1 aparece rápidamente en presencia de una concentración elevada de fósforo extracelular, asociado simultáneamente a una disminución marcada en la expresión génica de marcadores específicos de célula muscular lisa como es la α-actina de músculo liso (SM22). Los cambios fenotípicos vistos en el cultivo de CMLV con concentraciones altas de fósforo fueron regulados por Pit-1. El uso de ácido fosfonofórmico (PFA), un inhibidor específico de cotransportadores Na-P, prácticamente abolió la captación de fósforo en la CMVL. La expresión génica de osteocalcina y Cbfa-1 inducida por fósforo fue también inhibida por PFA en una manera dosis dependiente, validando la hipótesis que la entrada de fósforo a las CMLV y la posterior activación de genes osteogénicos es dependiente del cotransportador Na-P Pit-1. Estudios posteriores han determinado que el suero urémico puede contribuir in vitro a la calcificación de la pared vascular, independiente de los niveles de fósforo. Estudios en cultivos de anillos aórticos han demostrado que la calcificación puede ocurrir en ausencia de hiperfosfatemia. Vasos normales expuestos a concentraciones elevadas de calcio (1.8mM) y fósforo (3.8mM) no sufrieron calcificaciones. Sin embargo, la injuria mecánica del vaso resultó en extensa calcificación de la media de la pared vascular.
La hiperfosfemia induciría calcificación vascular en asociación con factores presentes en el suero urémico y en presencia de daño vascular, que es frecuente de encontrar en pacientes con insuficiencia renal.
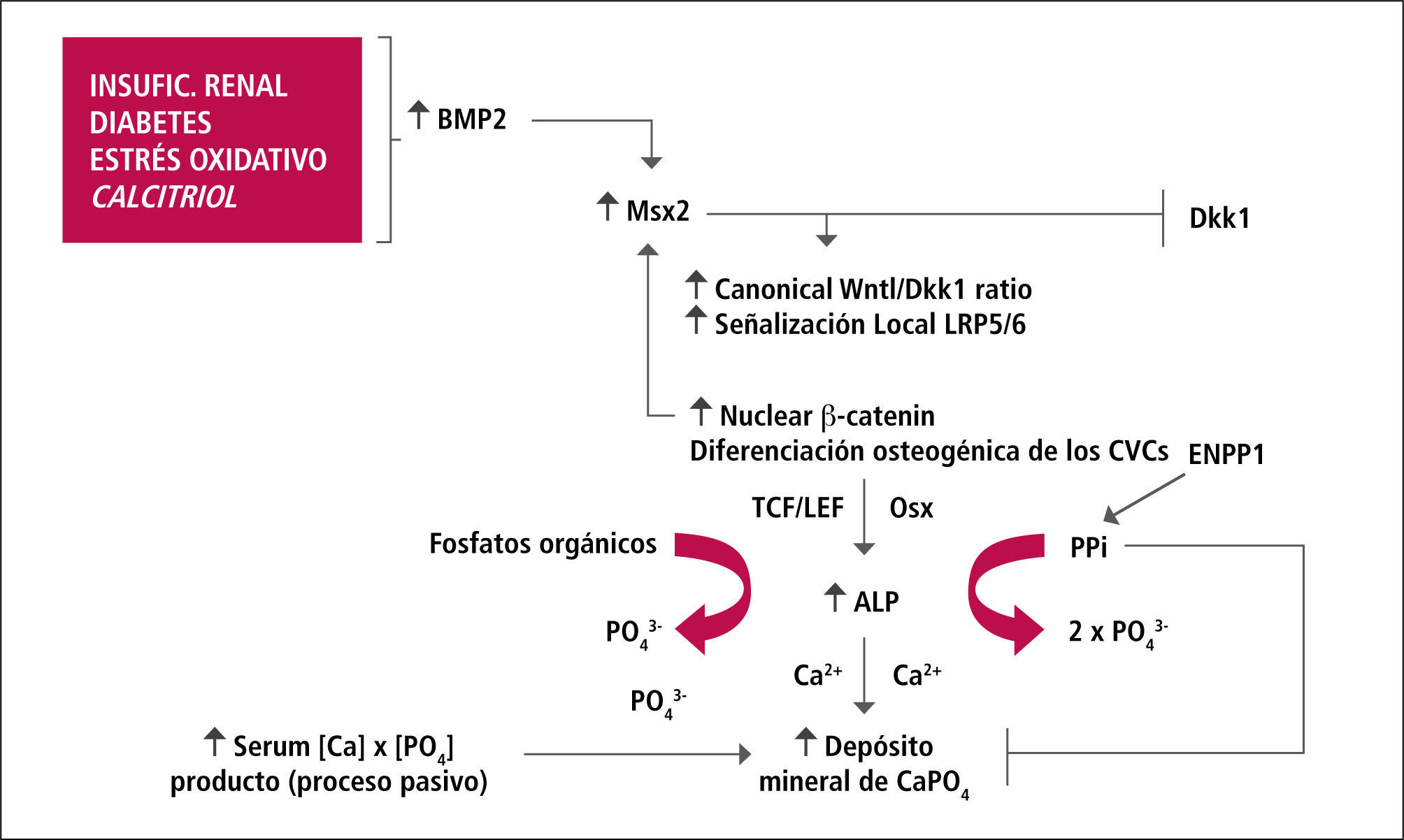
Genes del desarrollo temprano y calcificación vascularLas proteínas morfogenéticas de hueso (BMPs), llamadas así por sus propiedades osteoinductoras, fueron originalmente identificadas como proteínas que inducen formación ósea en sitios extraesqueléticos (22). Hay alrededor de treinta BMPs conocidas y, excluyendo a la BMP-1, todas ellas están estructuralmente relacionadas y pertenecen a los péptidos de la superfamilia del TGF-β (transforming growth factor-β). Actúan a través de la unión a un complejo heterodímero de receptores de transmembrana (receptor BMP I y II) que trimeriza después de la señal. La unión de una BMP a su receptor específico tipo II da como resultado la activación de los receptores tipo I. Esto provoca la fosforilación y la translocación nuclear de los factores de transcripción Smad (son factores de transcripción que una vez activados entran en el núcleo con el fin de formar complejos para el reconocimiento y regulación -activación o represión- de genes específicos) modificando así la tasa de transcripción de los genes blancos. Se ha descrito que inducen la formación ectópica de hueso.
De las diferentes BMPs, sólo BMP-2 y BMP-4 pueden inducir diferenciación osteogénica de la CMLV a través de la inducción de factores de transcripción Cbfal, osterix y MSX-2. Msx2 es requerido para la formación de hueso intermembranoso y Cbfa 1 es crítico para la diferenciación de osteoblastos, la formación de hueso endocondral y la neovascularización (23). Además BMP-2/BMP-4 contribuyen a la calcificación vascular al gatillar apoptosis de CMLV e inhibir el efecto de MGR El papel de BMP2 en la calcificación vascular está modulado (inhibido) por Matrix Gla Protein (MGP). BMP4 también se expresa en aortas dañadas. Actúa como agonista del receptor de BMP2.
Se ha planteado que el evento inicial en la calcificación vascular observada en uremia es la expresión de BMP-2, que gatillaría la transdiferenciación de la CMLV a célula símil a osteoblasto. Las señales en cascada gatilladas por BMP-2 pueden ser activadas por stress oxidativo mural y citoquinas inflamatorias sugiriendo que estas señales participan en la calcificación arterial que se observa también en pacientes diabéticos sin ERC y en modelos animales de síndrome metabólico (24). Se ha comunicado que una subpoblación de miofibroblastos localizados en la grasa de la adventicia aórtica y no en la túnica media, pueden elaborar esta respuesta BMP-Msx2 temprana. Estudios recientes muestran que señales osteogénicas vasculares tales como la señal paracrina Wnt/β-catenina, iniciada por acción de BMP-2-Msx2 de la adventicia, son concéntricamente dirigidas a la túnica media calcificada. Así, BMP-2 es un estímulo clave para la expresión de Msx2 y aumenta la señalización Wnt/ß-catenina. De este modo, BMP-2 puede activar una señal Wnt/β-catenina vascular que gatilla la mineralización osteogénica de progenitores vasculares. Diversos estudios experimentales han mostrado que mientras calcitriol induce calcificación vascular, paricalcitol –análogo de vitaminaD- inhibe la calcificación vascular en ratones urémicos. Ha sido demostrado que paricalcitol, a diferencia de calcitriol, inhibe la translocación nuclear de β-catenina al ser inhibida la señal Wnt, y por lo tanto, disminuye la diferenciación osteocondrogénica.
Las BMPs, al igual que otros factores de crecimiento, son reguladas tanto a nivel de expresión como de actividad, y los efectos de las BMPs pueden, entonces, ser modulados por grupos de polipéptidos que anta-gonizan la acción de ellas. Entre estos antagonistas se incluyen: noggin, follistatin, y gremlin (26).
Rol de Gremlin en el proceso de calcificación vascularGremlin juega un rol en diversos procesos durante el crecimiento, diferenciación y desarrollo embrionarios, mediante heterodimerización con BMPs, inhibiendo su capacidad de unirse a sus receptores. La expresión de gremlin ha sido demostrada durante el desarrollo del ducto pronéfrico en Xenopus y en varios modelos de patología humana. Su homólogo en rata, también llamado drm (down-regulated by v-mos) es una proteína de 184 aminoácidos altamente conservada durante la evolución y expresada preferencialmente en librerías de fibroblastos. Recientemente, in vitro, se ha caracterizado y clonado gremlin a partir de cultivos primarios de células mesangiales humanas. Se identificó aumento en la expresión de gremlin en células mesangiales humanas expuestas a medio rico en glucosa in vitro, y en modelos experimentales de nefropatía diabética in vivo. Además, estudios sugieren que la sobreexpresión de gremlin contribuye a la transdiferenciación de células epiteliales tubulares renales en cultivo hacia un fenotipo fibroblástico y a la formación de crescientes. Recientemente, se ha demostrado que gremlin se expresa en niveles muy bajos en osteo-blastos no estimulados, pero su expresión es aumentada por BMPs (26). El efecto estimulatorio de BMP-2 sobre gremlin fue dosis y tiempo dependiente y estuvo asociado con un aumento en los niveles de la proteína. Nuestro grupo identificó la sobreexpresión de gremlin en túnica media de arterias de pacientes urémicos y en modelo experimental de calcificación vascular en ratas urémicas tratadas con calcitriol (26).
Rol de Gremlin e inhibidores de la señalización Wnt/β-catenina en el remodelamiento óseoEje Hueso – VasoDurante el último tiempo se reconoce al osteocito como una célula fundamental en el remodelamiento óseo y constituye la población celular predominante en la trabécula ósea (90%). Es una fuente prominente de factores de remodelación ósea que incluye, entre otros, el receptor para el ligando activador de NF-kB (RANKL), osteoprotegerina y especialmente esclerotina, debido a que estos influencian la actividad osteoblástica y osteoclástica (27). La señalización Wnt/β-catenina es importante en la modulación de esta actividad, ya que se ha demostrado que regula actividad osteoclástica por afectar el cuociente RANKL/OPG en el osteocito. Por otro lado, la inhibición de la señalización Wnt/β-catenina disminuye la actividad osteoblástica. β-catenina es el componente clave de la señal Wnt canónica. En ausencia de Wnt, β-catenina forma complejo con APC y Axin, lo que facilita su fosforilación y posterior degradación por el sistema ubiquitina/proteosoma. Sin embargo, la interacción de Wnt con receptores FZD y LPR5/6 lleva a la degradación de Axin, lo que permite a β-catenina disociarse del complejo APC-Axin-GSK y traslocarse al núcleo y ejercer sus acciones genómicas permitiendo la diferenciación y proliferación osteoblástica (23, 28, 29). La señal Wnt es controlada estrechamente por varios grupos de reguladores negativos que interfieren con la unión del ligando con su receptor, tales como sFRPs y WIF-1, o por moléculas que antagonizan LRPs para bloquear la señalización Wnt, tales como Dickkopfs-1 (Dkk-1) y esclerotina (Figura 4).

Estudios en vasos de ratones urémicos sometidos a dieta con alto contenido de fósforo y que desarrollaron marcada calcificación aórtica, demuestran aumento significativo de la expresión génica de sFRPs en la aorta. Otros investigadores han demostrado, en similares modelos animales de calcificación vascular, un aumento de Dkk-1 circulante y a nivel local en el vaso calcificado (28). Gremlin, sFRPs y Dkk-1 afectan la formación ósea al inhibir la señalización Wnt/ β-catenina.
Puede plantearse entonces como hipótesis, que mientras el desarrollo de enfermedad ósea adinámica en el paciente con ERC favorece la calcificación vascular al afectar severamente el metabolismo de calcio y fósforo, el proceso de calcificación vascular genera por sí mismo un menor remodelamiento óseo y acentuación de enfermedad ósea adinámica. Esto se produce cuando el vaso en proceso de investigación genera moléculas que intentan frenar la diferenciación osteocondrogénica y la mineralización de la matriz extracelular, tales como gremlin, sFRPsy Dkk-1 (30, 31).
Así, estos mecanismos e interacción entre ellos pueden ayudar a explicar la correlación entre pérdida de masa ósea y calcificación vascular en población envejecida aún sin enfermedad renal (32).
El autor declara no tener conflictos de interés, con relación a este artículo.