



Los avances de la medicina nos enfrentan a nuevos desafíos como es el caso de los síndromes hereditarios que predisponen al desarrollo del cáncer. El modelo clásico del médico especialista que trata un tumor específico de su paciente, debería ser cambiado por el del “equipo de salud que asesora a una familia”. El diagnóstico de una enfermedad hereditaria tiene un fuerte impacto sicológico y emocional en cada uno de los miembros de la familia ya que implica adaptarse a cambios en el estilo de vida, adherirse a pautas de vigilancia activa mediante imágenes, exámenes de sangre y controles médicos periódicos e incluso en ciertos casos deberán someterse a cirugías de carácter profiláctico. En vista a este nuevo desafío, las instituciones están viendo en la necesidad de desarrollar e implementar Programas de Riesgo Alto Oncológico conformado por un equipo interdisciplinario, que permita acoger y apoyar a estas familias. En este artículo se describen las características de un programa de alto riesgo oncológico y la experiencia de un centro que ha implementado este programa.
Advances in medicine face new challenges, such as the hereditary syndromes that predispose to the development of cancer. The classic model of the specialist doctor who treats a specific tumor of his patient, must be changed by the “health team that advises a family”. The diagnosis of an inherited disease has a strong psychological and emotional impact on each family member as it involves adapting to changes in lifestyle, adhering to active surveillance guidelines through imaging, blood tests and periodic medical checks and even in some cases must undergo prophylactic surgeries. In view of this new challenge, institutions are seeing the need to develop and implement High-Risk Programs in Oncology, which is made-up of an interdisciplinary team, which allows them to host and support these families. This article describes the characteristics of a high-risk program in oncology and the experience of a center that has implemented this program.
El desarrollo de un Programa de Riesgo Alto Oncológico (PRAO) surge de la observación de profesionales relacionados con el área oncológica de la existencia de familias en que varios miembros desarrollan tumores en su mayoría a edades tempranas. Lo que llamó fuertemente la atención es que en estas familias se desarrollan tumores 20 ó 30 años antes de lo habitual y más aún, hay casos donde una misma persona desarrolla múltiples tumores primarios.
El PRAO, también conocido como Programa de Tumores Hereditarios (PTH) tiene los siguientes objetivos:
- 1.
Identificar familias en riesgo.
- 2.
Educar a familiares sobre los síndromes hereditarios.
- 3.
Ofrecer una vigilancia del cáncer de acuerdo a los factores de riesgo, incluyendo el riesgo genético.
- 4.
Pesquisar mutaciones genéticas de la línea germinal (heredadas).
Estos objetivos permitirán reducir la mortalidad por cáncer mediante la detección precoz e incluso su prevención, tanto a nivel primario como secundario. Permiten también diagnosticar el síndrome hereditario, idealmente identificando el gen afectado. Para poder llevar a cabo estos objetivos se ha hecho necesario establecer un programa que esté constituido por varios componentes, partiendo con la creación de un “registro” seguido de la formación de un “equipo de trabajo” que se capacite continuamente, y “protocolos de trabajo” para implementar un trabajo organizado y transversal.
EL REGISTROEl registro puede estar a cargo de un coordinador, que puede ser algún profesional de la salud o el asesor genético, quien al evaluar al caso índice le solicitará su consentimiento para ser incorporado y se le entregará una encuesta, se construirá la genealogía, se evaluará el riesgo y se realizará vigilancia activa del mismo y su familia.
EncuestaEl contenido de la encuesta dependerá del síndrome al que pertenece el caso índice, sin embargo, lo ideal es tener una línea similar dentro de la institución. Es importante considerar sólo información que está respaldada con informes anátomo-patológicos de biopsias o informes médicos. Es importante registrar otras afecciones y co-morbilidades diferentes al cáncer y hábitos en el estilo de vida (alimentación, deportes, controles médicos, exámenes de rutina, medicamentos), ocupación y si eventualmente ha estado expuesto a elementos carcinogénicos (contaminantes industriales, radiación). También es relevante incluir la percepción subjetiva de riesgo que tiene el paciente. Si el asesor genético utiliza modelos de riesgos, se debe considerar incluir en la encuesta información pertinente como edad actual, si tiene ascendencia judío-ashkenazi, edades de menarquia y menopausia, número de partos, entre otros, para evaluar el riesgo. También se recomienda incluir información de médicos e instituciones donde se ha tratado al paciente y sus familiares.
GenealogíaLa genealogía es fundamental ya que con ella se puede analizar a la familia en su conjunto y determinar la línea o rama familiar de los miembros que han desarrollado el cáncer, su parentesco, edad, tipo de tumor, entre otros y emitir recomendaciones en cuanto a qué gen o genes serían los más apropiados para analizar. La genealogía permitirá también, identificar aquellos familiares de alto riesgo. Para ello se debe incluir información de al menos 3 generaciones consecutivas o hasta los familiares de 2° grado de todos los afectados (lo que involucre la mayor información) y también de todos los familiares sanos. Es importante recabar con precisión las fechas de nacimiento, edad y motivo de fallecimiento de los miembros de ambas ramas familiares. Si fue por cáncer, registrar órgano o tejido afectado, tipo histológico, edad de diagnóstico, número de tumores benignos como malignos, si hay segundos tumores o recidivas. También, patologías asociadas u otras enfermedades genéticas.
Para diseñar las genealogías existen diferentes programas computacionales siendo uno de los más utilizados “CLINICAL (Pedigree and Clinical Data)” de Progeny 1. También existen otros sitios web como el de The Cleveland Clinic que permite utilizar on-line “Cologene” el cual está diseñado especialmente para registrar y dibujar genealogías de familias con cáncer colorrectal hereditario 2.
Evaluación de riesgoAlgunos de los PRAO utilizan modelos de riesgo como Gail, Claus, Tyrer-Cuzick, BRCAPRO, BOADICEA, entre otros, para realizar cálculos empíricos como herramientas de apoyo para categorizar a los pacientes en aquellas que hay sospecha de síndrome mama/ovario. Estos modelos son muy utilizados en Estados Unidos, pero es discutible su aplicación en poblaciones latinoamericanas, ya que el modelamiento se ha hecho con datos de pacientes estadounidenses. Existen sitios donde se puede utilizar programas que contienen estos modelos como por ejemplo CancerGene3.
Una vez concluida esta etapa, se discutirá en un comité ampliado y se decidirá si se justifica el realizar un estudio genético o solo mantener una vigilancia de acuerdo al riesgo familiar observado. Es importante recalcar que hay criterios de riesgo familiar para solicitar los estudios genéticos, sin embargo, puede ser que una familia en particular no reúna los criterios hoy para justificar el estudio genético, pero en un corto plazo si sean candidatos (desarrollo de tumores en otros miembros). Por esta situación, se debe enfatizar el papel de incorporar a las familias en riesgo en un registro y mantener una comunicación fluida entre ellos y el programa.
Vigilancia Activa de familias del registroEl seguimiento tanto del caso índice como de los familiares de riesgo alto es clave y para ello es ideal contar con una herramienta informática que permita generar alarmas cuando los pacientes índices y/o familiares en riesgo no se han realizado sus estudios de seguimiento o no han acudido a sus controles. Progeny a través de su programa FHQ (Family History Questionnaire) permite que el paciente llene y actualice su información personalmente, mediante un acceso que le llega vía correo electrónico, también permite preguntarle al paciente datos de sus familiares y si autoriza a contactarlos, por lo que este programa permite también identificar a los familiares de riesgo alto.
Es importante considerar que existirán dos grupos de familiares de riesgo alto:
- 1.
Aquellos donde se ha identificado una mutación en la familia que explica el mayor riesgo a desarrollar un síndrome dado y;
- 2.
Aquellos en que no, pero que la genealogía muestra una clara agregación de familiares con cáncer.
En el primero, las recomendaciones de vigilancia y/o recomendaciones de cirugías profilácticas van a estar dadas por las guías clínicas en las cuales el equipo interdisciplinario se fundamente, como por ejemplo las guías de la NCCN o ATA, entre otras. En aquellos familiares donde se haya descartado la presencia de una mutación identificada previamente en un familiar, es decir con resultado Verdadero Negativo, es importante aconsejar la adhesión a los programas de prevención estándar, nutrición saludable y balanceada y actividad física, mejorando los hábitos en el estilo de vida, para prevenir el desarrollo de tumores esporádicos.
En el segundo, las recomendaciones irán acorde a las características de la genealogía, es decir, los tipos de cáncer que se manifiesten en una familia dada y según el criterio y experiencia del equipo del PRAO.
EQUIPO DE TRABAJOPara desarrollar un PRAO se necesita contar con un equipo de trabajo interdisciplinario. Es necesario que esté liderado por un coordinador del programa quien se encargue de definir, por ejemplo: vías de derivación para la atención de los pacientes, algoritmos de trabajo de los diferentes comités. Además, debe: coordinar la elaboración e implementación de protocolos de forma homogénea entre los distintos comités, apoyar actividades de formación continua en genética y cáncer y en síndromes de cáncer hereditario de todos los miembros del programa, coordinar la elaboración del material educativo para pacientes y coordinar y apoyar al asesor genético en el desarrollo de sus actividades.
También es importante que algún miembro del equipo desarrolle la función de asesor genético, cuyas funciones son: realizar evaluación de riesgo a individuos y familiares que consultan espontáneamente o que son derivados, detectar individuos con sospecha de síndrome de cáncer hereditario o agregación familiar, llevar un registro de los pacientes y familias que consultan y mantener información actualizada, coordinar la toma de muestra para la realización de estudios genéticos para aquellos que tienen indicación, disponer de material educativo para los pacientes. Para ello es fundamental que este Coordinador del registro y/o asesor genético sepa construir un árbol genealógico, conocer los diversos patrones de herencia y saberlos identificar en el árbol genealógico de las familias atendidas, calcular los riesgos que posee cada familia de presentar un cáncer hereditario, conocer y saber identificar los síndromes de predisposición familiar que se conocen a la fecha, saber comunicar al paciente los posibles problemas genéticos a los que se enfrenta con un lenguaje accesible al entendimiento de la población general, ser empático, conocer los límites éticos y legales que implica conocer el estado genético de una determinada persona de una familia, respetando la confidencialidad de la información genética.
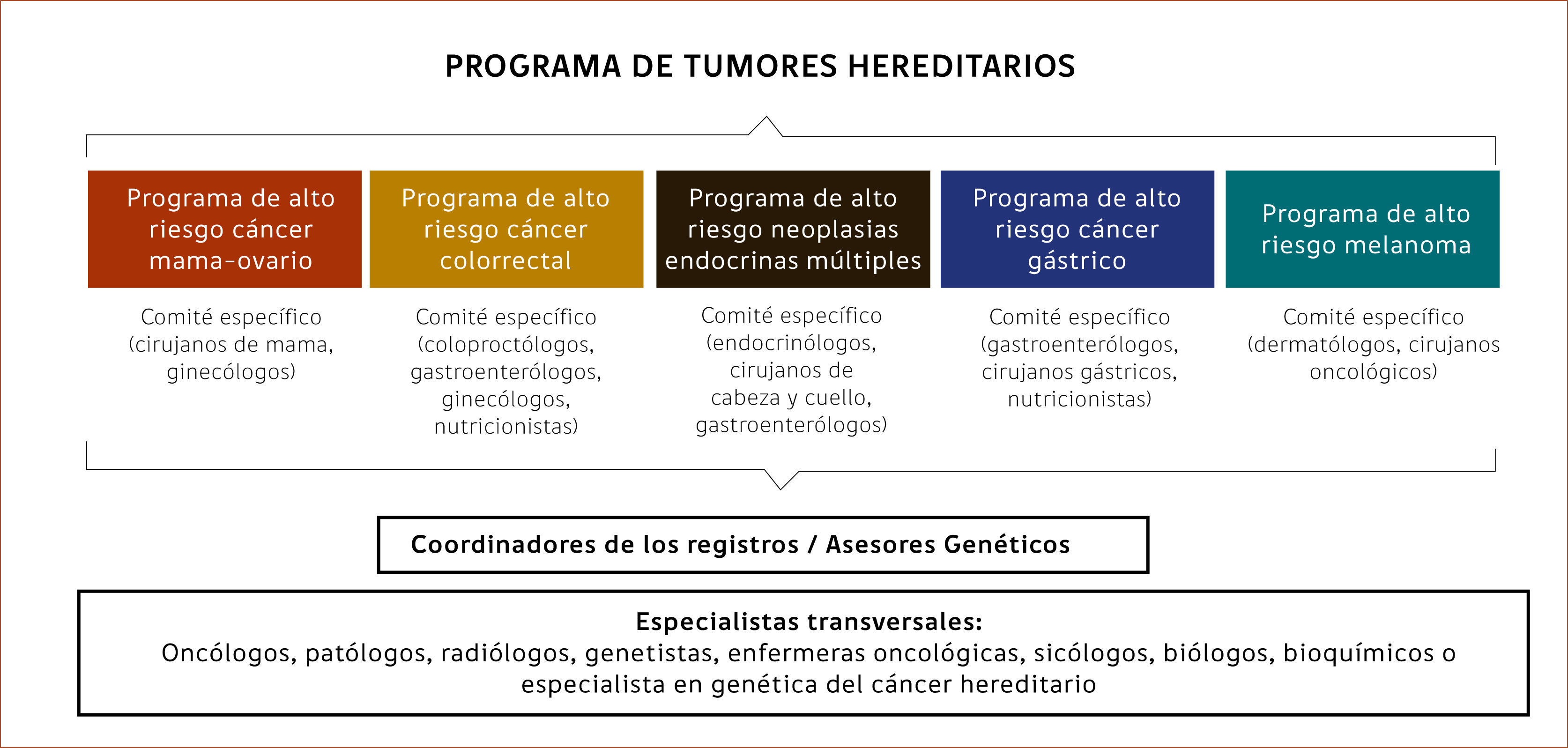
Es ideal que el equipo esté inserto dentro de un Centro Oncológico e involucre al menos a los especialistas de los síndromes hereditarios de mayor impacto oncológico: asesor genético, oncólogos, cirujanos de mama, ginecólogos, coloproctólogos, gastroenterólogos, cirujanos digestivos, cirujanos endocrinos, endocrinólogos, urólogos, neurocirujanos, genetistas, pediatras, entre otros. Junto a ellos es necesario que se incorporen profesionales de la salud que entreguen un apoyo transversal como radiólogos, patólogos, psico- oncólogos, bioquímicos, enfermeros u otro profesional que esté a cargo del registro, que en muchas ocasiones cumple también el rol de asesor genético y apoyo administrativo. El PRAO idealmente debe abarcar distintos síndromes que se pueden ordenar a su vez en distintos registros que están organizados por un comité o equipo interdisciplinario y especialidades médicas según sean los tipos de tumores (Esquema 1). Todos deben cumplir una serie de protocolos de forma transversal para lograr una uniformidad en los procesos y asegurar el cumplimiento de pautas de seguridad y resguardo de la información.

También debe haber una comunicación fluida entre los distintos equipos ya que se pueden presentar casos en los cuales se requiera realizar interconsultas. Todo ello es fundamental para entregar la mejor atención a las familias.
Por lo tanto, el desarrollo de programas de este tipo requiere el esfuerzo mancomunado de diferentes especialistas que trabajen integradamente. Para ello se hace necesario mantener un espacio de reuniones regulares para discusión de nuevas familias incorporadas al registro, resultados de estudios genéticos y puesta al día de nuevos conocimientos. En estas reuniones se debieran discutir los protocolos transversales y presentar casos complejos que involucren múltiples especialidades.
Cada Comité evaluará las nuevas familias que han sido referidas para el programa, la decisión de los estudios que se realizarán, las conclusiones sobre resultados de los estudios genéticos y la pauta de asesoría que será entregada. De esta forma se deberían establecer Comités de Mama, Colon, Endocrino, entre otros. que se debieran reunir en forma autónoma de acuerdo al flujo de pacientes que manejen. Se recomienda que en estos Comités debe haber un encargado y un secretario de actas quien debe enviar los acuerdos tomados al resto de los miembros y realizar un registro de asistencia junto al acta.
Otros componentes que influyen en el buen desarrollo del programa son los médicos derivadores y campañas de prevención, quienes deben dar respuesta a temas generales sobre cáncer hereditario, reconocer a pacientes que presentan riesgo superior al poblacional y derivarlos al PRAO, realizar vigilancia activa de pacientes, conocer los criterios de riesgo de cáncer hereditario, conocer protocolos de derivación para pacientes de riesgo moderado y alto, disponer de información médica sobre síndromes y factores de riesgos, mantener una formación continua.
El Comité Ético de la institución por su parte, debe revisar y aprobar protocolos y documentos que se utilizan en el PRAO, de acuerdo a las leyes vigentes y a las buenas prácticas clínicas. Además, debe supervisar el correcto almacenamiento de la información de los pacientes y apoyar al comité interdisciplinario cuando requiera consultar situaciones especiales que necesiten de una aprobación del comité de ética.
Capacitación continua del equipoUn aspecto trascendente es la formación de asesores genéticos quienes deberán informar al caso índice y a los familiares en riesgo. Por ejemplo, en EE.UU. existen varias universidades que imparten la maestría de “Consejero/a Genético”, dictado para enfermeras o profesionales del área de la biología 4 y tienen una duración de 2 años, lo que significa un gran apoyo para los PRAO. En Latinoamérica no existe este programa de formación, pero se han desarrollado Diplomados que entregan las herramientas básicas a los profesionales interesados para que puedan adquirir conocimientos que les permitan construir y desarrollar programas de alto riesgo 5.
Por lo tanto, el programa debe contar con profesionales que sean capaces de interpretar los cambios genéticos y entregar un asesoramiento que involucra compromisos que impactarán la calidad de vida de personas asintomáticas. Recordemos que con excepción del caso índice (primer miembro de la familia que se acerca al programa, habitualmente por presencia de un tumor sintomático), el resto de los familiares son “sanos” y asintomáticos. Para esto se debe contar con un registro activo en el que se incorpore información de acuerdo a los cambios de la familia y/o el desarrollo de nuevos tumores. El manejo de esta información requiere de la máxima confidencialidad para lo cual se debe contar con protocolos de ingreso y transferencia de información a los distintos miembros del equipo de salud con quienes interaccionen estos pacientes, todos elementos que seguiremos desarrollando más adelante.
Finalmente, queremos mencionar una iniciativa pionera en Latinoamérica, que consistió en la creación de un Diploma de Asesoramiento Genético en Cáncer Hereditario para profesionales de la salud pertenecientes a unidades oncológicas. Este trabajo fue realizado gracias el esfuerzo diferentes profesionales pertenecientes a Clínica Las Condes, Universidad del Desarrollo, Pontificia Universidad Católica de Chile, Universidad de Chile, Kaiser Permanente (USA) y University of Southern California (USA). El diplomado consiste en 220 horas de estudio, 180 de las cuales son a través de una plataforma e-learning (a distancia) y 40 horas en un taller presencial (cinco días). Es así como el 30.4% de los estudiantes fueron internacionales pertenecientes a Colombia 7, Perú 2, Argentina 2 y República Dominicana 1. Un total de 39 alumnos se han titulado entre los años 2013 y 2015, que incluye 26 médicos, 9 enfermeras, 2 PhD, 1 tecnólogo médico y 1 biólogo. El aumento en la demanda por esta formación, es debido principalmente a la necesidad de mantenerse actualizados en el conocimiento y a la disponibilidad de la información que actualmente poseen los pacientes quienes demandan hoy una mejor atención clínica.
Protocolos de trabajoTodo programa tiene que estar organizado y deben existir protocolos de procedimiento, así como documentación que debe mantenerse actualizada y según las exigencias de la institución.
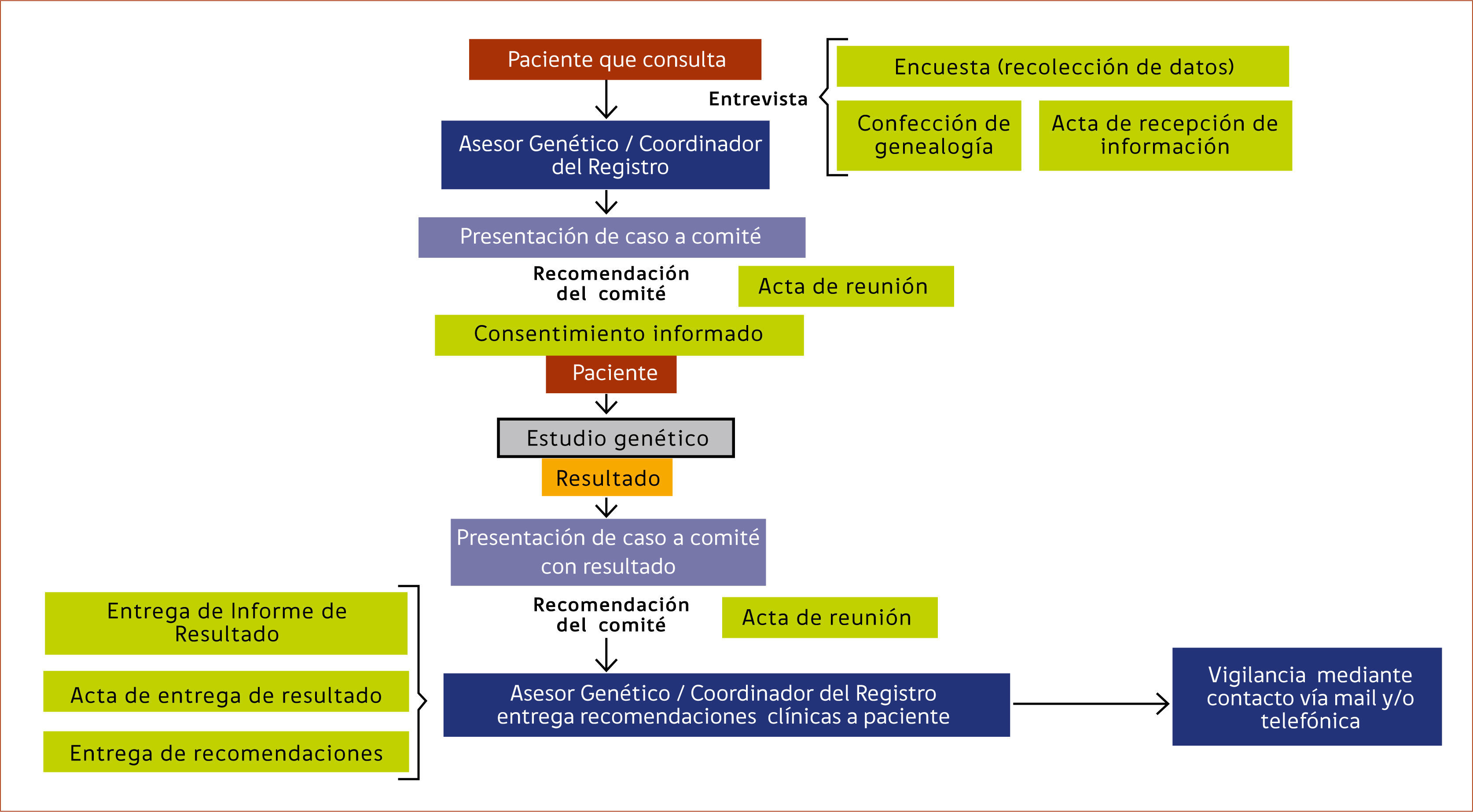
El desarrollo de flujogramas de trabajo permite ordenar y sistematizar el trabajo del equipo. En el esquema 2 se muestra el algoritmo utilizado en el asesoramiento genético de los pacientes que consultan en nuestro centro, indistintamente del comité por el cual es evaluado.

Documentos de información para pacientes: Es muy importante contar con material de apoyo visual, como una carpeta con elementos explicativos de la evaluación de riesgo y consejería genética pre-test, así como de folletos que se entregan a los pacientes para educar y difundir información sobre los síndromes hereditarios, redactados con un lenguaje simple y sintético 6.
Actas y respaldo de la información: Se recomienda redactar documentos de respaldo como parte de protocolizar el programa, entre ellos se aconseja contar con actas de entrega de información, actas de reunión de comités al presentar cada caso y actas de entrega de resultados.
Consentimiento Informado: El consentimiento informado debe considerar: Título, Introducción, Propósito, Beneficios, Procedimiento, Riesgos, Privacidad y Confidencialidad, Participación, Costos, Explicación de los posibles resultados del estudio genético y su interpretación, Limitaciones del estudio, uso de la muestra, contacto de los responsables del programa, acta de consentimiento y acta de revocación. El acta de revocación es un derecho del paciente que le permite retractarse del estudio durante el proceso de análisis de su muestra.
Emisión de recomendaciones: La emisión de recomendaciones dependerá de cómo lo desee realizar cada equipo en base a las guías clínicas utilizadas como referencia. Algunos de ellos prefieren entregar por escrito a los pacientes las recomendaciones que han sido emitidas por el comité. Sin embargo, otros equipos prefieren solamente emitir las recomendaciones de forma verbal y derivar al paciente nuevamente a su médico tratante de origen. También se debe considerar derivar a apoyo psico-oncológico al paciente y familiares como parte de un programa integral.
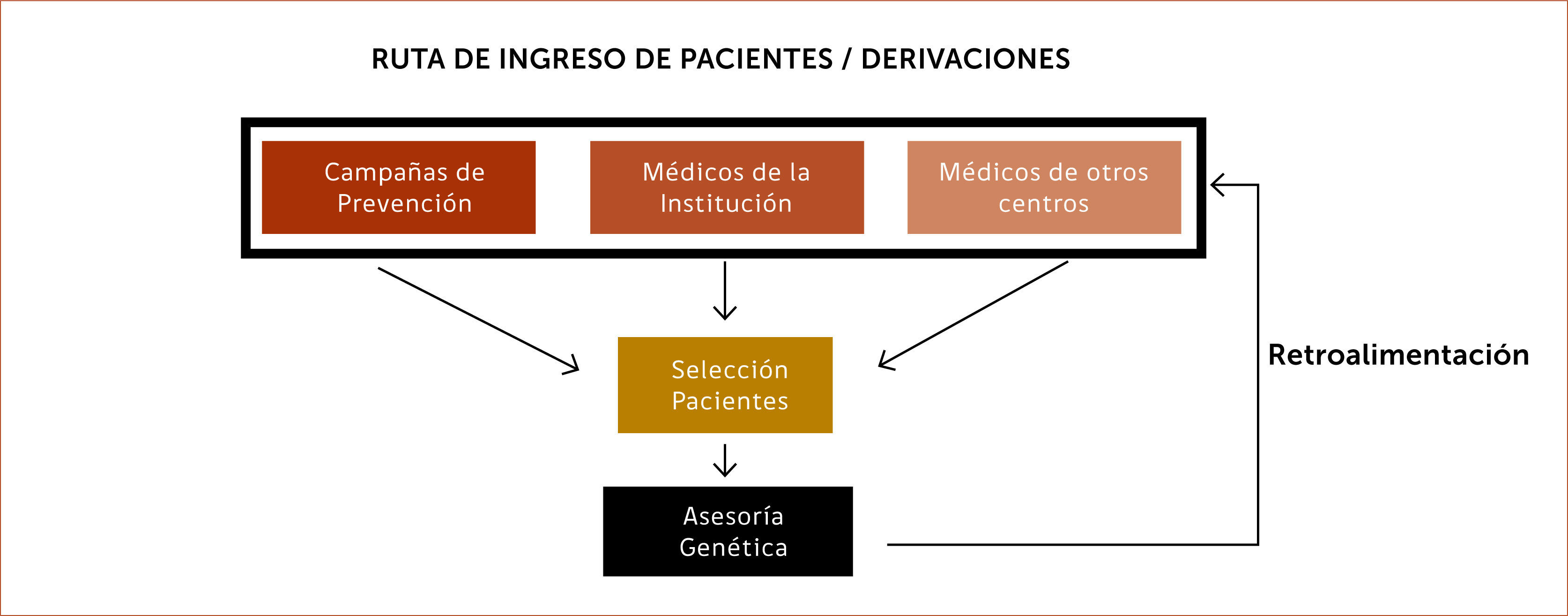
Estrategias de pesquisa de familiares de alto riesgo oncológico: Existen diferentes estrategias para pesquisar familias de riesgo alto oncológico. Una de ellas es, por ejemplo, incluir encuestas dentro de campañas de prevención estándar de cáncer 7,8 que permitan obtener información de aquellas familias y ofrecerles un asesoramiento genético (Esquema 3).

También ha resultado exitoso incorporar este tipo de encuestas a pacientes que se acercan a las unidades de imágenes de la mama o bien a procedimientos de colonoscopía o, cuando se acercan a consulta con uno de los especialistas que integran el equipo de riesgo alto oncológico por primera vez. Un ejemplo de formas de aplicar encuestas es como se implementó en Nueva Jersey, EE.UU., donde las pacientes que van a realizarse mamografías contestan un cuestionario mientras esperan a través de una Tablet. Los datos ingresados permiten aplicar inmediatamente modelos de riesgo y categorizar a las pacientes 9.
El diseño de la o las encuestas dependerá de la forma de organización de los equipos encargados de cada tipo de síndrome, o bien, se puede diseñar una encuesta general y aplicar a todo paciente que consulta en la institución por primera vez y luego derivarla al equipo de riesgo alto según cumpla criterios.
Otra forma es a través de las mismas páginas web de las instituciones donde la persona puede llenar una breve encuesta “on-line”. Si cumple con ciertos criterios la persona es contactada y se le ofrece una evaluación de riesgo más completa 10,11.
También las instituciones han logrado pesquisar familias de riesgo alto al ofrecer estudios genéticos que son financiados por proyectos de investigación ya sea con fondos estatales o privados. Estos proyectos son difundidos a través congresos nacionales y a nivel de la comunidad impartiendo charlas educativas 12–15.
Países como Brasil han realizado incluso planes a nivel municipal de pesquisa, realizando visitas puerta a puerta y derivando las familias de riesgo alto desde la atención primaria a centros de salud especializados 16. En Argentina por su parte, se creó en el año 2011 un Plan Nacional de Tumores Familiares y Hereditarios del Instituto Nacional del Cáncer y a través de políticas de gobierno trabajando de forma conjunta con el Ministerio de Salud han desarrollado un manual para la práctica clínica en el Asesoramiento Genético en Oncología, el cual resume distintos puntos clave que deben ser consideradas en el asesoramiento 17. Por otro lado, en Chile, si bien no existen políticas públicas que desarrollen este tipo de programas, existen instituciones privadas que están estableciendo y difundiendo estos Programas de Riesgo Alto Oncológico (PRAO) 18,19.
EXPERIENCIA DE LA IMPLEMENTACIÓN DEL PROGRAMA DE RIESGO ALTO ONCOLÓGICO EN NUESTRO CENTRO1.PROGRAMA DE ALTO RIESGO DE CÁNCER COLORRECTAL (CCR) HEREDITARIO
Registros Cáncer Colorrectal HereditarioAproximadamente el 10% de los casos de CCR son hereditarios, con una herencia autosómica dominante, cuyo origen es debido a una mutación de la línea germinal en genes previamente identificados. Entre los principales síndromes se encuentran el Síndrome de Lynch (5%) y la Poliposis Adenomatosa Familiar (1%), además de otros síndromes muy infrecuentes (<1%) como el Síndrome de Peutz-Jeghers, el Síndrome de Cowden y la Poliposis Juvenil.
Nuestro interés en el CCR hereditario nace el 2002, luego de identificar una familia con Síndrome de Lynch (HNPCC1) con cuatro hermanos angustiados y resignados a desarrollar la enfermedad, al igual que sus múltiples familiares fallecidos por CCR antes de los 50 años 20. En esos momentos, en el país existía una carencia de los estudios genéticos y de la aplicación de recomendaciones clínicas para la detección precoz de la enfermedad. Por tal motivo, el año 2003 postulamos a fondos concursables del gobierno chileno (FONDECYT) para la caracterización genético-molecular del Síndrome de Lynch y de la Poliposis Adenomatosa Familiar. Gracias a este importante apoyo económico para la investigación, pudimos durante los años 2004-2006: implementar el análisis de inestabilidad microsatelital e inmunohistoquímica en los tumores colorrectales, e implementamos la búsqueda de mutaciones puntuales en los genes MLH1, MSH2 y APC 13,14,21,22. Durante los años 2008-2012, logramos el apoyo económico de Cleveland Clinic y Clínica Las Condes para continuar investigando sobre las causas genéticas involucradas en el cáncer colorrectal hereditario. Para ello, implementamos el estudio de deleciones/duplicaciones mediante MLPA (Multiplex ligation-dependent probe amplification) y se amplió la búsqueda de mutaciones a nuevos genes candidatos como MSH6, PMS2, MUTYH, STK11, BMPR1A, PTEN y SMAD4 15,23,24. Si bien, se logró aumentar la tasa de pesquiza de mutaciones con la inclusión de nuevos genes y con la implementación de esta nueva tecnología, en los años venideros ocurrió un gran avance tecnológico con el uso de la secuenciación masiva, la que permitió estudiar paneles de genes. Estos paneles son especialmente recomendables cuando existe más de un gen involucrado en la etiología de un síndrome, en aquellos pacientes cuyo estudio del tumor no permite definir un gen candidato, o cuando se sospecha más de un síndrome hereditario (colon y mama/ovario). La costo-efectividad de estos paneles está dada por el estudio de hasta 25 genes relacionados a CCR al mismo tiempo, con el uso de programas bioinformáticos que aceleran los análisis, con un tiempo de respuesta de tres semanas y a menor costo; a diferencia del estudio secuencial, que implicaba evaluar primero un gen, luego otro y así sucesivamente, a través de distintas metodologías, requiriendo una mayor utilización de horas hombre. Actualmente, todos los estudios de los casos índices son derivados a un panel de genes según el síndrome que se sospecha, en tanto que los diagnósticos de los familiares son realizados en nuestro laboratorio.
A la fecha, nuestro registro cuenta con 100 familias sospechosas del Síndrome de Lynch, con un total de 212 personas registradas. De estas familias, 30/100 (30%) tienen mutación identificada en los genes reparadores, según la siguiente distribución: 19 en MLH1, 7 en MSH2, 2 en PMS2 y 2 con deleción de EPCAM 14,24,25. Además, el registro cuenta con 108 familias con poliposis colónica, con un total de 250 personas registradas. Las familias están clasificadas de la siguiente manera: 94 Poliposis Adenomatosa Familiar, 10 Síndrome de Peutz-Jeghers, 2 Poliposis Juvenil, 1 Síndrome de Cowden y 1 Poliposis hiperplásica. En total, 45/108 (41.7%) tienen mutación de la línea germinal identificada: 35 familias con Poliposis Adenomatosa Familiar con mutación en APC 13, 8 familias con Síndrome de Peutz-Jeghers con mutación en STK11 15, 1 familia con Poliposis Juvenil con mutación en SMAD4 y 1 familia con Cowden con mutación en el gen PTEN.
Interesantemente, seis mutaciones en el Síndrome de Lynch y diez mutaciones en los síndromes polipósicos, no han sido descritas en otros estudios, demostrando la relevancia de evaluar diferentes poblaciones debido a las distintas influencias genéticas como es el caso de la nuestra que involucra una mezcla de poblaciones amerindia y europea, principalmente española. Todas las familias con mutación identificada han recibido las recomendaciones clínicas reportadas en la literatura y actualmente utilizamos las guías de la National Comprehensive Cancer Network (NCCN).
2.PROGRAMA ALTO RIESGO DE MAMA/OVARIO
Registros Cáncer de mama y Alto RiesgoCáncer de mama y cáncer de ovario son causas importantes de morbilidad y mortalidad. Datos del Ministerio de Salud señalan un número aproximado de 4000 nuevos diagnósticos de cáncer de mama por año 26. Sin embargo, ello no refleja la verdadera realidad de Chile ya que no existen registros adecuados de cáncer en el sector privado de Salud.
El año 1995, el Centro Integral de la Mama de Clínica Las Condes (CIM), inicia un registro de pacientes atendidas por cáncer de mama que ha permitido conocer sus características epidemiológicas, efectuar su seguimiento y evaluar sus programas terapéuticos. El registro ha aportado datos relevantes para la auto-evaluación del CIM y el desarrollo de investigación clínica, lo que ha contribuido a la mayor comprensión de esta patología. El análisis del seguimiento de pacientes registrados ha permitido conocer datos epidemiológicos que complementan los registros de instituciones públicas.
Después de 20 años de desarrollo, el Registro del CIM ha sido recientemente regularizado desde el punto de vista ético y científico. Se genera un proyecto para su revisión y actualización titulado “Validación y Análisis del Registro Histórico de Pacientes con Cáncer de Mama atendidos en el Centro Integral de la Mama (CIM) de Clínica Las Condes, desde el año 1995”, para incluir de manera prospectiva a todas las pacientes. Actualmente, el registro de cáncer de mama acumula 2056 pacientes con Cáncer de Mama diagnosticados y tratadas en Clínica las Condes.
En forma paralela, el año 2008 se inicia en el CIM el Programa de Alto Riesgo de Cáncer de Mama/Ovario, que selecciona y registra individuos y familias de mayor riesgo de desarrollar la enfermedad estableciendo protocolos de estudio y estrategias de vigilancia y prevención.
Los portadores de mutaciones germinales (heredadas) asociadas a mayor predisposición al cáncer constituyen un grupo de muy alto riesgo para desarrollar cáncer de mama y ovario, Estos individuos pertenecen a familias con el síndrome de cáncer de mama y ovario hereditario que afecta aproximadamente a 10% de los pacientes con cáncer de mama y 10-15% de pacientes con cáncer de ovario y se caracteriza por el desarrollo de cáncer de mama y ovario a edades más tempranas 27. La identificación de portadores de mutaciones es muy relevante ya que se les puede ofrecer estrategias específicas de prevención y de reducción de riesgo permitiendo además un mayor entendimiento de la fisiopatología de la enfermedad. El Programa de Alto Riesgo de Cáncer de Mama y Ovario en CLC registra familias que están en riesgo y provee asesoría genética para la prevención del cáncer. Permite, además, estudiar las mutaciones genéticas presentes en la población chilena.
Un registro de Alto Riesgo permite evaluar el desarrollo del programa y sus alcances dentro y afuera de la institución, además apoya la investigación científica para llegar a conocer las características de las mutaciones hereditarias de nuestra población 28–31. Como la población chilena es genéticamente heterogénea, las mutaciones genéticas presentes pueden ser diferentes a las de otras poblaciones 32. Es por ello la importancia de estudiar estas mutaciones y evaluar su correlación con el fenotipo de la enfermedad.
Hasta la fecha se han registrado en el programa 651 mujeres de alto riesgo de cáncer de mama/ovario que corresponden a 576 casos índices más 75 familiares contactados y estudiados. Del análisis de cada caso clínico en el Comité en Alto Riesgo, que funciona semanalmente, surgen recomendaciones de estudio y de seguimiento. Entre las recomendaciones de estudio está la realización de estudios genéticos de los genes BRCA 1-2 y paneles de genes según los antecedentes familiares de cada caso. Se han realizado 293 estudios genéticos, de ellos 237 estudios completos y 56 análisis moleculares dirigidos al estudio de una determinada mutación. De los 237 estudios completos hubo 28 positivos para mutaciones patogénicas, 183 negativos, 16 variantes inciertas y 10 estudios aún pendientes. De los 56 familiares en que se determinaron mutaciones específicas, 25 fueron positivas para la mutación, 30 negativas y una variante incierta. De 40 mutaciones encontradas, 25 corresponden a mutaciones de genes BRCA 1-2, dos mutaciones en PALB2; una en CHEK2 y una en RAD51D.
Es necesario continuar avanzando en el estudio genético de familias de muy alto riesgo para ofrecerles las mejores opciones de vigilancia y prevención.
3.PROGRAMA ALTO RIESGO DE NEOPLASIAS ENDOCRINAS MULTIPLES
Registros Neoplasias endocrinas múltiplesLas Neoplasias Endocrinas Múltiples (NEM) son un conjunto de síndromes hereditarios que involucran el desarrollo de tumores en distintas glándulas endocrinas. Si bien en su mayoría estos tumores son de comportamiento benigno y de baja prevalencia, existen tumores agresivos con una importante morbi-mortalidad y con alta incidencia en personas jóvenes 33.
Los pacientes con sospecha de alguno de estos síndromes se caracterizan por presentar tumores en al menos 2 o más glándulas a lo largo de su vida. Dependiendo de las características y glándulas afectadas, se han identificado principalmente dos tipos de NEM: tipo 1 y tipo 2. El primer síndrome (NEM tipo 1) se explica principalmente por mutaciones inactivantes en el gen supresor de tumor MEN1 y se caracteriza por la presentación de tumores paratiroideos que producen hiperparatiroidismo primario y adenomas pituitarios o tumores entero-pancreáticos; por su parte, el segundo (NEM tipo 2), está dado por mutaciones activantes en el oncogén RET y se caracteriza por el desarrollo de hiperplasia de las células C la glándula tiroides (parafoliculares) que evoluciona siempre hasta progresar finalmente en cáncer medular del tiroides (CMT), acompañado en algunos casos de feocromocitoma e hiperparatiroidismo primario 33.
En Chile, alrededor del año 2000 se analizaron las primeras familias chilenas con sospecha de NEM1 y NEM2 mediante un proyecto de investigación encabezado por el Dr. Nelson Wohllk 34. Estos estudios finalizaron con el término del proyecto. Luego de algunos años, un grupo de médicos especialistas planteó la necesidad de implementar el estudio del gen RET en nuestra institución y derivar a aquellos pacientes diagnosticados con cáncer medular de tiroides (CMT), para poder identificar aquellas familias con NEM tipo 2 y así poder descartar aquellos casos que eran esporádicos. Esto culminó en la creación del registro de pacientes con sospecha de NEM en nuestro centro en el año 2012. Es por ello que, a mediados de ese mismo año, el laboratorio de Oncología y Genética Molecular de Clínica Las Condes desarrolló e implementó este estudio genético como servicio clínico. Para levantar el programa se organizaron reuniones semanales de revisión de casos y se derivaron los primeros pacientes. A la fecha, el registro contiene 68 pacientes diagnosticados con CMT y que han sido derivados para estudio del gen RET desde distintos centros de todo Chile. Esto ha permitido identificar a la fecha a 12 familias con mutación y estudiar a más de 40 familiares con el fin de identificar si son también o no portadores de mutación. Se identificaron mutaciones en miembros de la familia asintomáticos y que luego del diagnóstico molecular se les recomendó volver con sus médicos tratantes, para que ellos les indiquen la vigilancia adecuada. La creación y mantención del registro de estos pacientes nos ha permitido mantener contacto con ellos y saber por ejemplo, que aquellos que salieron portadores se les indicó entre otros, exámenes de imágenes, permitiendo diagnosticar tempranamente nódulos hiperplásicos de células C en la tiroides y en un caso lamentablemente ya un cáncer medular de tiroides en una persona joven. La indicación del estudio genético de RET a tiempo, sin duda es fundamental ya que existen en varios casos portadores asintomáticos incluso en edades bien avanzadas debido a la penetrancia incompleta y expresividad variable de esta enfermedad.
Con la misma necesidad que surgió de implementar el estudio genético de RET, se solicitó la incorporación de estudio del gen MEN1 y actualmente se tienen un registro de 35 pacientes con sospecha de NEM1 y de ellos se ha identificado mutación en 10 de ellos, permitiendo a la fecha estudiar a 10 familiares, algunos aún asintomáticos.
Finalmente, dado los avances día a día sobre nuevos genes que explican el mayor riesgo a desarrollar manifestaciones clínicas similares actualmente se está extendiendo la implementación a otros genes como AIP que explica casos de hiperparatiroidismo familiar aislado (FIHP) y CDKN1B para sospecha de casos de NEM tipo 4 (pacientes con hiperparatiroidismo, tumores hipofisiarios y tumores adrenales y gonadales), con el fin de ofrecer un servicio más completo para los médicos tratantes que requieren de apoyo molecular para el diagnóstico más preciso de sus pacientes.
Actualmente el programa cuenta con estos registros de familias estudiadas, gracias a un protocolo de registro que fue aprobado por el comité de ética de la institución y con la finalidad de tomar todas las medidas que sean necesarias para resguardar la confidencialidad de los pacientes y sus familiares.
Los autores declaran no tener conflictos de interés en relación a este artículo.