



El cáncer es una de las principales patologías que afectan a la población a nivel mundial. La introducción de estrategias de detección precoz y mejoras en la terapia del cáncer han permitido en países desarrollados disminuir su incidencia y mejorar la sobrevida de los pacientes. Este progreso ha sido generado por el vertiginoso avance en la investigación básicoclínica durante las últimas décadas. La medicina traslacional ha llevado el conocimiento del laboratorio a la práctica clínica y requiere de la férrea interacción entre investigadores básicos y médicos. La descripción de los genes, proteínas y el microambiente que caracteriza a los tumores entrega información específica para facilitar el diagnóstico y pronóstico y entregar el tratamiento más eficaz, aplicando una medicina personalizada. Conocer en mayor detalle los procesos biológicos que caracterizan esta enfermedad permitirá acelerar la introducción de nuevas técnicas moleculares a la práctica clínica habitual, mejorando las estrategias de prevención y manejo de los pacientes.
Cancer is one of the major diseases that concern population worldwide. In developed countries, early detection strategies and improvement in cancer therapy have decreased its incidence and improved the survival of cancer patients. This progress has been reached by the vertiginous advance in basic and clinical research during the past decades. Translational medicine has taken the knowledge from laboratory to clinical practice and it requires the strong interaction between basic researchers and clinicians. The description of the genes, proteins and the microenvironment that characterize the tumors gives specific information that may improve the diagnosis and prognosis, and help to prescribe the most effective treatment, practicing a personalized medicine. To know the biological process that characterizes this disease will accelerate the acquisition of new molecular techniques to clinical practice, improving prevention strategies and patient management.
El cáncer es una de las principales patologías que afectan a la población a nivel mundial. Según la Organización Mundial de la Salud (OMS), el 2008 se diagnosticaron 12,7 millones de nuevos casos y 7,6 millones de personas murieron de cáncer, lo que representa un 13% de todas las defunciones a nivel mundial (1). En nuestro país, el cáncer representa la segunda causa de muerte tras las enfermedades del sistema circulatorio (2), lo que se aproxima a lo observado en países desarrollados, como Estados Unidos. Según datos del Ministerio de Salud, el 2010 fallecieron 23.136 personas de cáncer (135,3/100.000 habitantes), lo que representa un 23,6% dentro de todas las causas de muerte (2).
En países desarrollados, la introducción de estrategias de detección precoz y las mejoras en la terapia contra el cáncer han permitido disminuir, en algunos casos, la incidencia de cáncer y además, mejorar la sobrevida de los pacientes afectados. Mejorar en estas áreas ha sido posible gracias al incremento en el número de proyectos de investigación llevados a cabo en las últimas dos décadas. La investigación científica ha permitido la descripción de las causas y mecanismos fisiopatológicos de esta enfermedad.
Sin embargo, para que este conocimiento sea de utilidad, debe ser llevado a la práctica clínica, lo que es una tarea ardua y requiere de la interacción entre investigadores básicos y médicos. Esto ha dado origen a la medicina traslacional.
El cáncer : Una enfermedad celularConocer y describir los mecanismos fisiopatológicos del cáncer es una tarea compleja. El término “cáncer” agrupa entidades clínicas de diverso origen, como cáncer de mama, neuroblastomas, osteosarcomas o leucemias entre otras. A pesar de las distintas manifestaciones clínicas de esta enfermedad, ha sido posible generar un consenso en ciertos principios comunes que se observan en las distintas entidades clínicas (3), cuya complejidad se ha ido descifrando gracias a los avances tecnológicos.
Ya en el comienzo del estudio de esta enfermedad, en 1838, se describió que el tejido canceroso estaba conformado por células con morfología alterada, y se postuló que la causa de esta enfermedad yacía en lesiones celulares (4). Actualmente, el cáncer es considerado como un desorden de células que se dividen anormalmente, lo que conduce a la formación de agregados que crecen dañando tejidos vecinos, se nutren del organismo y alteran su fisiología. Además, estas células pueden migrar e invadir tejidos lejanos, donde encuentran un nicho apropiado para continuar su crecimiento originando una metástasis (5) que en muchas ocasiones es la causa de muerte de los individuos afectados.
Durante los últimos años, el desarrollo de nuevas tecnologías ha permitido conocer diversos aspectos de la fisiología celular. El estudio de las células cancerosas tanto a nivel celular, molecular, metabólico y genético ha permitido mejorar de manera significativa el manejo de los distintos aspectos clínicos de esta enfermedad, como el crecimiento tumoral, invasividad y metástasis y entregando antecedentes que permitan predecir la sensibilidad a distintos tipos de terapia (6). En el ámbito clínico, esto se ha traducido en la introducción de biomarcadores tumorales (moléculas que se expresan en niveles anormales en ciertos tipos de cáncer y pueden ser detectadas para diagnosticar o analizar la evolución de una enfermedad) y la identificación de potenciales blancos terapéuticos. Por lo tanto, la aplicación de conocimientos básicos de fisiopatología celular se ha traducido en mejoras en las estrategias preventivas, diagnósticas, terapéuticas y pronósticas para los pacientes afectados o en riesgo de cáncer (7).
¿Cómo se originan las células tumorales ?El proceso por el cual las células normales se transforman en cancerosas se denomina carcinogénesis. La comprensión de este proceso se logró principalmente por el desarrollo de técnicas de estudio genético. Mediante estas, se estableció que la transformación progresiva de células normales a derivados altamente malignos se originaba en alteraciones en el material genético (mutaciones) (3, 5). Estas mutaciones le confieren a una célula la capacidad de dividirse a una tasa mayor que su cohorte y generar una descendencia que conserva esta mutación (clones). Posteriormente, las células hijas acumulan subsecuentes y diversas mutaciones que permite generar distintos clones. Estos presentan mayores capacidades de sobrevida y crecimiento, ventajas proliferativas respecto de su contraparte normal que permite generar un clon neoplásico persistente (5). Normalmente, las células del sistema inmune son capaces de eliminar a estas células tumorales, en un proceso denominado inmunovigilancia tumoral. Sin embargo, algunos de estos clones pueden adquirir nuevas capacidades que les permiten evadir estos mecanismos de control y se desarrolla una neoplasia (8; 9).
El rol de las alteraciones genéticas en la carcinogenesis fue puesto de manifiesto al descubrir en el genoma humano, genes homólogos a genes retrovirales relacionados previamente con el desarrollo de tumores. En células humanas normales estos genes se denominaron protooncogenes y se relacionan con el crecimiento y proliferación de las células normales. Cuando se encuentran mutados se denominan oncogenes y su mutación es de tipo dominante, es decir, sólo es necesario que uno de los alelos sufra una mutación para que la proteína que codifica, gane funcionalidad. Esto generalmente se traduce en aumento de sobrevida y proliferación (10).
Sin embargo, estos no son los únicos genes que explican el desarrollo tumoral. La descripción por parte de Knudson, de un modelo de 2 hits en el desarrollo del retinoblastoma asociado a la mutación del gen RB1, llevó indirectamente al descubrimiento de los genes supresores de tumores, que controlan la proliferación, reparación celular y apoptosis (muerte celular) (10). Knudson describió que en individuos afectados por retinoblastomas se produce una primera mutación en la línea germinal (primer hit) que inactiva uno de los alelos del gen RB1, dejando el otro alelo funcional, en un estado de heterocigosis, lo que disminuye a 50% la cantidad de proteína funcional. Para que se genere un tumor, debe ocurrir una segunda mutación somática en el alelo normal de RB1 (segundo hit) que lleva a la pérdida de la expresión de la proteína. Por lo tanto, para que se desarrolle la enfermedad, ambos alelos deben estar mutados, por lo que la mutación es de tipo recesiva. En este caso, las mutaciones de los genes supresores de tumores se traducen en una pérdida de su función, de las proteínas que codifican y por lo tanto, una falla en los mecanismos de control y reparación internos de la célula, permitiendo su proliferación y crecimiento descontrolados, además de la acumulación de nuevas mutaciones (3). El mecanismo por el cual se pierde la copia normal del gen se ha denominado pérdida de heterocigosis o LOH (por su nombre en inglés: Loss Of Heterozygocity) que es la principal forma de silenciamiento de genes supresores de tumor. Las mutaciones que explican la LOH son variadas y generalmente afectan grandes segmentos cromosómicos, por lo que se pueden pesquisar mediante técnicas moleculares que detectan la pérdida de marcadores cromosómicos aledaños al gen de interés, en particular de secuencias denominadas microsatélites. En general, un tumor con alta incidencia de LOH se relaciona con un pronóstico desfavorable (11).
Se presume que en una célula normal ocurren diariamente alrededor de 20.000 eventos que dañan el ADN y cerca de 10.000 errores de replicación (12). Las células poseen mecanismos complejos y a veces redundantes para la reparación de alteraciones o daño en el ADN, en los que están involucrados los genes de reparación del ADN. Existen alrededor de 153 genes que participan directamente en la reparación del ADN, cuyos principales mecanismos incluyen la reparación de mal pareamiento (o missmatch), reparación por escisión de base o nucleótido, unión de extremos no homólogos y recombinación homóloga. Algunos ejemplos de estos genes son BRCA1 y 2 (relacionados con el cáncer de mama y ovario), y MSH2, MLH1 y MSH6 (relacionados con cáncer colorrectal hereditario no poliposo). Cuando ocurren mutaciones en estos genes, la disfunción de las proteínas que codifican hace a las células más sensibles a agentes que dañan el ADN y a la adquisición y acumulación de nuevas mutaciones que favorecen la carcinogenesis. Algunos individuos son portadores de mutaciones heterocigotas en estos genes, lo que se asocia a una mayor susceptibilidad de desarrollar distintos tipos de cáncer (13).
Las mutaciones de los genes responsables de la carcinogenesis pueden ser heredadas o ser adquiridas de novo (o mutaciones somáticas) generalmente producto de la exposición a sustancias del ambiente (carcinógenos) o agentes biológicos (virus oncogénicos), o ser heredadas. En las últimas dos décadas se han descrito más de 50 síndromes de susceptibilidad a cáncer de alta penetrancia, ligados a la herencia de mutaciones en genes específicos. A pesar de que la prevalencia de estas mutaciones es baja, en la clínica ha representado un gran avance en términos de la introducción de estrategias preventivas a través de la evaluación de familias de alto riesgo (5, 10).
Para que estas mutaciones iniciadoras o promotoras de tumores logren persistir en una célula y dar origen a un clon tumoral, a nivel de la célula y su microambiente deben darse dos eventos fundamentales, que son comunes a todos los tipos tumorales: la inestabilidad genómica que favorece la adquisición de mutaciones y la inflamación tumorigénica.
1Inestabilidad genómica y mutacionesLa presencia y acumulación de las mutaciones responsables de la progresión tumoral está favorecida por un estado de inestabilidad genómica en las células tumorales. Esta es una característica común de la gran mayoría de los tumores que acelera la acumulación de cambios genéticos. Comúnmente, la inestabilidad genómica se manifiesta como grandes aberraciones cromosómicas y cambios en la ploidia, aunque también pueden observarse pequeños cambios a nivel nucleotídico, con inserciones, deleciones o sustituciones de nucleótidos. Las aberraciones cromosómicas ocurren temprano durante la transformación maligna, mientras que la inestabilidad genómica promueve la adquisición de capacidades que favorecen la progresión tumoral (14).
En células normales existen varios mecanismos que controlan la acumulación de mutaciones que ocurren de manera espontánea: la detención del ciclo celular, la reparación del ADN y la eventual destrucción de una célula muy dañada, mediante apoptosis. En este proceso participan las proteínas de los genes reparadores del ADN y los genes supresores de tumor y en particular, dentro de estas últimas, cumple un rol fundamental la proteína p53, denominada por esta importante función, el guardián del genoma. En general, las células tumorales acumulan mayor cantidad de mutaciones debido a que la tasa de mutaciones en ellas es mayor, producto de una mayor sensibilidad a agentes mutagénicos y/o por fallas en uno o más puntos de la maquinaria de control de la integridad genética ocasionadas por mutaciones en genes supresores de tumor o reparadores del ADN, por lo que la célula defectuosa no es destinada a senescencia o apoptosis (15).
Existen ciertas condiciones hereditarias que favorecen el desarrollo de mutaciones. En el síndrome de Lynch, los pacientes heredan genes reparadores del ADN mutados. Como producto de la falla en estas proteínas, las secuencias génicas no son preservadas correctamente durante la replicación y se generan nuevos fragmentos microsatélites, lo que genera un estado de inestabilidad microsatelital (MSI), la que puede ser pesquisada como marcador. Estos pacientes presentan un status basal de mutaciones en sus células que puede predisponerlas al desarrollo de diversos tipos de tumores (colon, endometrio, ovario, estómago, entre otros) (16).
Concomitante con la falla en los mecanismos de control, la pérdida del ADN de los telómeros (secuencias de ADN localizadas en los extremos de los cromosomas) es otra fuente de inestabilidad genómica, lo que explica alteraciones en el cariotipo de las células tumorales como amplificación o deleción de segmentos de cromosomas (14).
A pesar de que las mutaciones varían entre distintos tipos de tumores, su cantidad y presencia en el genoma tumoral ha demostrado que la inestabilidad genómica es inherente a los tumores. Esta finalmente, aumenta la probabilidad de que ocurran mutaciones en oncogenes que generan las capacidades que mejoran su sobrevida (12).
2Inflamación tumorigénicaLos tejidos normales del organismo están compuestos por distintos tipos de células. En el caso de los tumores, interactúan con las células cancerosas un conjunto de células que colaboran al crecimiento tumoral, dando el soporte funcional y nutricional, estableciendo lo que se ha denominado el microambiente tumoral. Dentro de estas, se encuentran fibroblastos anormales, células endoteliales y del sistema inmune innato y adaptativo (17).
Las células del sistema inmune son las principales responsables de la inmunovigilancia tumoral y eliminación de los clones tumorales. Sin embargo, durante este proceso se produce un estado de inflamación crónica mediado principalmente por macrófagos y mastocitos que infiltran el tumor y que producen factores que promueve el crecimiento tumoral en todas sus etapas (18). Por una parte, la inflamación promueve la iniciación tumoral al generar un estrés genotóxico, que favorecen nuevas mutaciones; participa en la promoción al inducir la proliferación tumoral y a la progresión tumoral al incrementar la producción de nuevos vasos sanguíneos (angiogenesis) alrededor del tumor y la invasión tisular al favorecer la extravasación celular, lo que facilita el desarrollo de metastasis (19, 20). Los factores generados por las células inmunes como factores proangiogénicos y de crecimiento, enzimas modificadoras de la matriz extracelular y otras señales son capaces de inducir las capacidades de las células tumorales y se han descrito como eventuales blancos terapéuticos (15, 19).
Capacidades comunes de las células tumorales : Entender para vencerA pesar de las diversas entidades clínicas agrupadas como cáncer, es posible identificar ciertas características comunes de las células tumorales que permiten entender esta enfermedad y el desarrollo de nuevas estrategias clínicas comunes para su manejo (3).
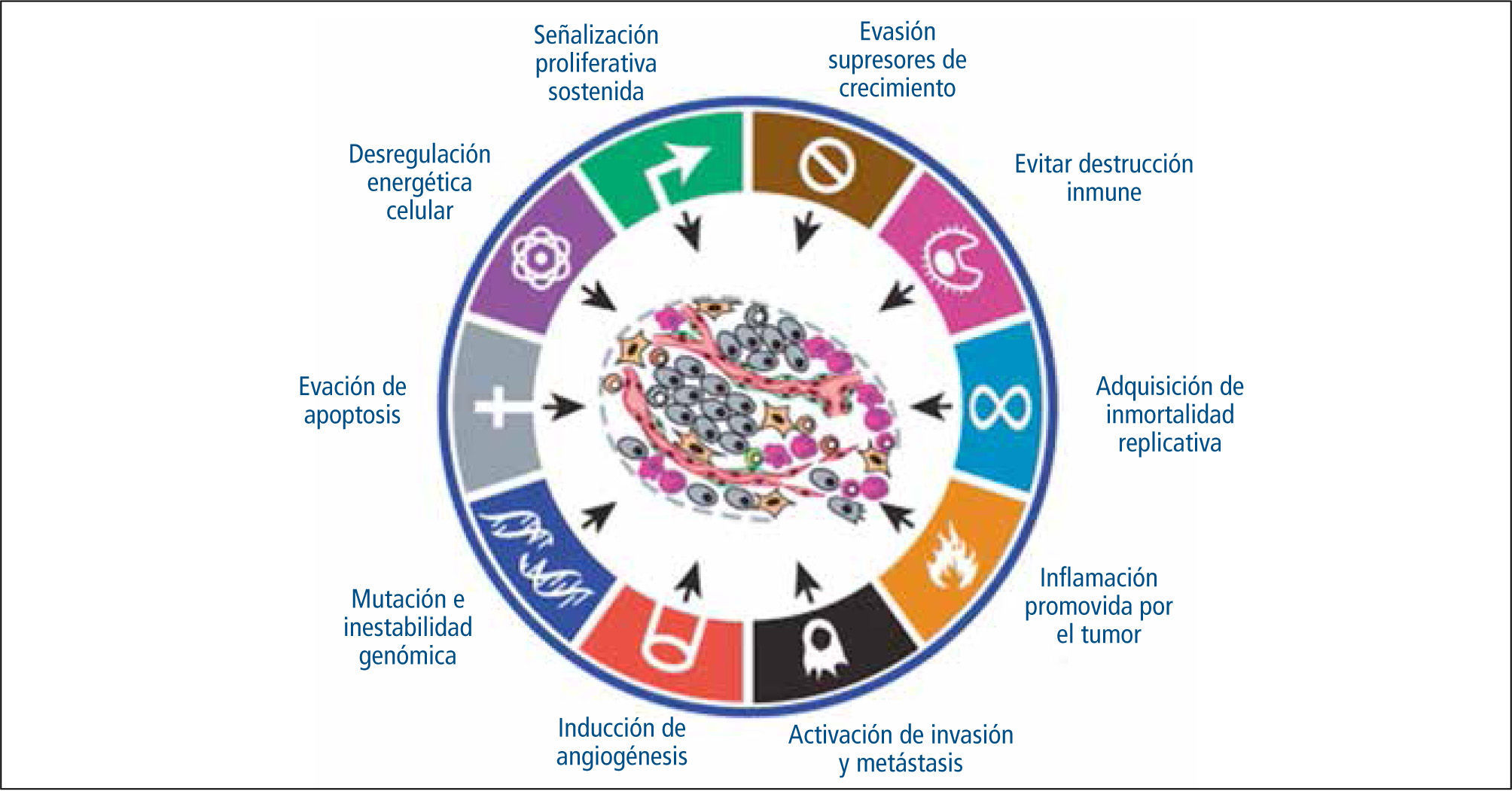
Hanahan y Weinberg el año 2000, tras un exhaustivo análisis de la literatura publicada, describieron 6 características que son compartidas por las células tumorales:
- 1.
Independencia de señales de crecimiento.
- 2.
Insensibilidad a estímulos que inhiben el crecimiento.
- 3.
Invasividad y metástasis.
- 4.
Invasividad y metástasis.
- 5.
Potencial ilimitado de replicación.
- 6.
Angiogenesis sostenida (3).
Estas características son adquiridas en los diversos tipos celulares por distintos mecanismos y eventos durante el proceso carcinogénico. El año 2011, estos autores describieron 2 nuevas características: reprogramación del metabolismo energético y evasión de la destrucción inmune, lo que concluye en 8 características intrínsecas de las células tumorales (Figura 1). Su desarrollo se ve favorecido por la inestabilidad genómica y la inflamación las fomenta (15).

A continuación se describen en mayor detalle estas características y algunos de sus alcances clínicos.
1) Independencia de señales de crecimientoUna de las características más evidentes de las células tumorales es su alta tasa de proliferación. A diferencia de los tejidos normales, se pierde la homeostasis que regula cuidadosamente la entrada al ciclo de división celular y por ende, el número de células y la arquitectura y función del tejido.
Normalmente, las células son estimuladas por señales externas de proliferación (principalmente factores de crecimiento) que activan receptores presentes en la superficie celular, los que contienen típicamente dominios de tirosina quinasa intracelular. Este evento permite la activación consecutiva de vías de señalización intracelular que regulan la progresión a través del ciclo celular, el crecimiento de la célula, favoreciendo la supervivencia celular (15).
En las células tumorales se observan mutaciones en los genes involucrados en estas vías de señalización (en general oncogenes) que mantienen activa la proliferación de manera anormal por uno o varios de los siguientes mecanismos: sintetizan o estimulan en sus células vecinas la síntesis de ligandos similares a los factores de crecimiento que estimulan a la célula tumoral; expresan nuevos receptores de membrana que responden a las señales proliferativas presentes en el entorno y/o sintetizados por la misma célula; aumentan la expresión de receptores tirosina quinasa en la superficie, haciéndose hipersensibles; presentan mutaciones en los receptores de superficie o en las proteínas de la vía de señalización río abajo del receptor, lo que las mantienen constitutivamente activas; o presentan mutaciones en las proteínas encargadas de la retroalimentación negativa que permite la atenuación de la señal proliferativa (15).
La descripción de estas alteraciones ha permitido la identificación de una serie de blancos terapéuticos que han logrado controlar el crecimiento de las células tumorales. La leucemia mieloide crónica se produce por una mutación en los genes Bcr/Abl que genera una proteína mutante tirosina quinasa constitutivamente activa que sólo se expresa en células tumorales. Esto se usó como blanco terapéutico y permitió el desarrollo de la primera terapia dirigida exclusivamente contra células tumorales. Esta droga, el imatinib (de la familia de inhibidores de tirosina quinasas) tras la inhibición de la proteína anormalmente activa, gatilla la apoptosis exclusivamente de las células que expresan la proteína mutante. Otras drogas de esta familia son gefitinib y erlotinib contra el receptor EGF y sunitinib que inhibe los receptores para FGF, PDGF y VEGF (21).
Mutaciones en la vía de señalización MAPK (proteína quinasas activadas por mitógenos) que se encuentra río abajo de los receptores tirosina quinasa, explican el desarrollo de una serie de tumores (22). El oncogen RAS (HRAS, NRAS y KRAS), que pertenece a esta vía, es el oncogen más frecuentemente mutado en canceres humanos (33%), por lo que su inhibición representa un gran desafío en la terapia del cáncer. Esta se ha logrado a través de la inhibición farmacológica de proteínas efectoras de RAS, en particular Raf y MEK, con fármacos ya aprobados como sorafenib y vemurafenib y otros muchos en estudio. Otra vía de señalización comúnmente alterada en tumores es la vía PI3K-Akt-mTOR, relacionada con la proliferación y sobrevida, en la que generalmente se pierde la función de la proteína PTEN (supresora de tumor). Actualmente, se encuentran disponibles para la inhibición de mTOR las drogas temsirolimus y everolimus y otras se encuentran bajo estudio (23).
2) Insensibilidad a estímulos que inhiben el crecimientoLa tasa de proliferación aumentada de las células tumorales también se encuentra favorecida por la evasión de los mecanismos de regulación negativa de la división celular, controlados por los genes supresores de tumor. Estos genes actúan a distintos niveles, limitando el crecimiento tumoral y la proliferación (15).
Existen distintas vías de regulación que actúan de manera concertada y complementaria, lo que en ciertos casos pueden suplir la ausencia de alguna de ellas tras mutaciones que ocasionan la pérdida de función. Dentro de estas, dos genes han sido ampliamente estudiados ya que sus mutaciones son comunes a varios tipos de tumores: RB (asociada a Retinoblastoma) y TP53.
La proteína RB integra señales externas e internas de proliferación, decidiendo si la célula está en condiciones de progresar en el ciclo celular, en particular en la transición G1/S. Además regula la expresión génica de proteínas que participan en la mitosis. Mutaciones en esta proteína se observan en un 100% de los retinoblastomas y en menor proporción en cáncer de pulmón, vejiga, hígado, esófago (24).
Por otra parte, p53 censa distintas formas de stress intracelular (por ejemplo, daño en el ADN, cantidad de nutrientes), activando dos vías respuestas principales: arresto en el ciclo celular en la transición G1/S para permitir la reparación del ADN o la apoptosis cuando el daño es demasiado o irreparable. P53 se encuentra mutado en alrededor del 50% de los tumores humanos. Su mutación se puede originar por la exposición a agentes cancerígenos como radiación, químicos y virus como el papiloma humano (HPV). Por otra parte, la presencia de una proteína p53 funcional en un tumor es un predictor de buena respuesta a terapia en pacientes que reciben tratamientos que inducen apoptosis (25).
Algunos pacientes heredan un alelo mutado y otro funcional del gen p53 (son heterocigotos), lo que los predispone al desarrollo de diversos tumores en la edad adulta, condición denominada síndrome de Li-Fraumeni. Estos pacientes tienen 25% más de probabilidad de desarrollar un tumor maligno, el que se produce cuando ocurre la mutación del alelo normal del gen (el segundo hit, según la teoría de Knudson). Este síndrome se relaciona con alta incidencia de cáncer de mama, tumores cerebrales, leucemia, sarcomas y carcinoma adrenocortical. La mutación de p53 gatilla la tumorigénesis al perderse su actividad supresora de tumor (25, 26).
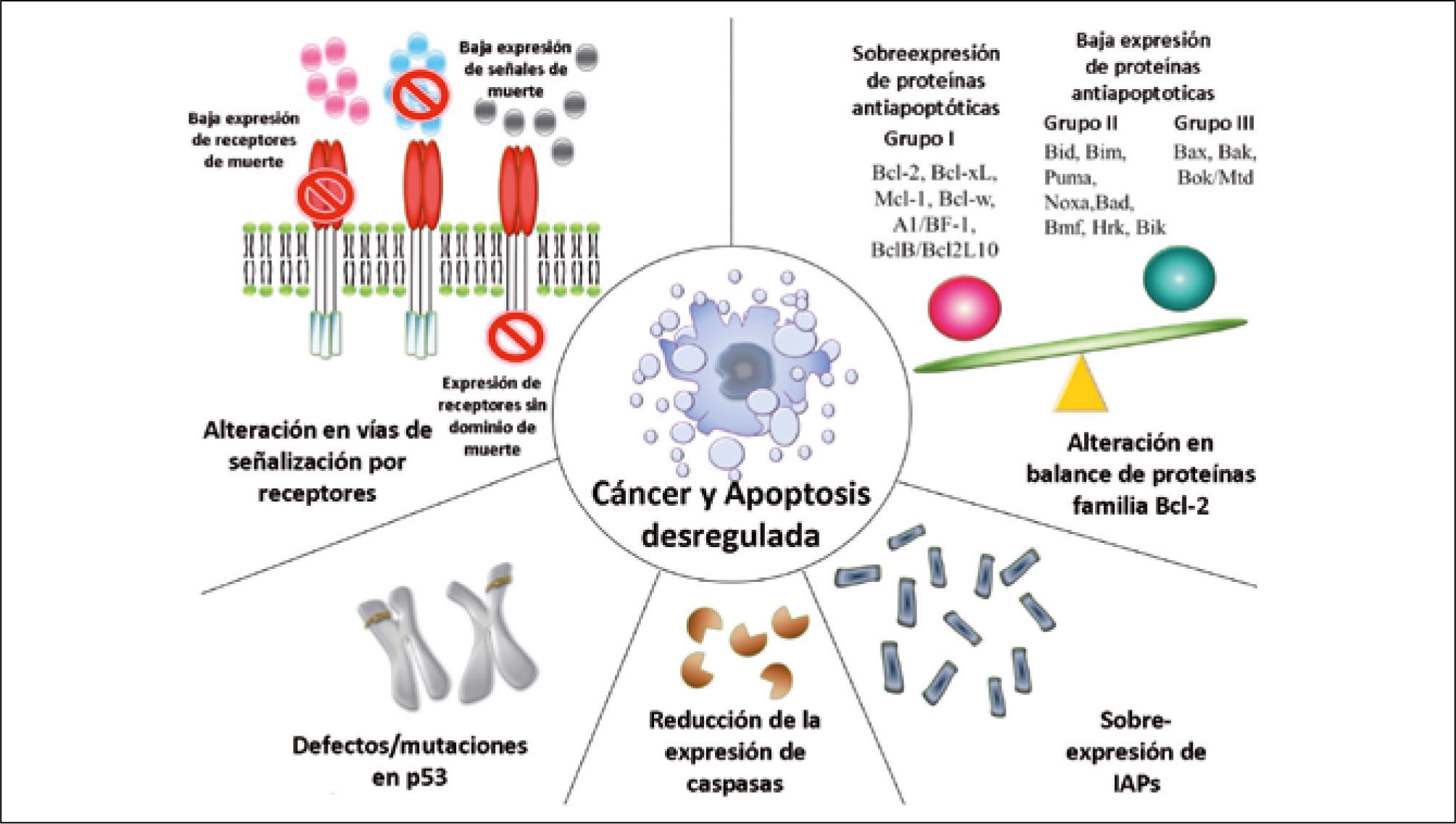
3) Evasión de apoptosisEl crecimiento de los tumores no sólo depende de cuanto se dividen las células que lo componen sino que también está condicionado por la tasa de muerte celular. La disminución del número de células ocurre principalmente por apoptosis, un mecanismo normal que mantiene la homeostasis tisular. La apoptosis es gatillada por diversos estímulos, los que confluyen en vías moleculares comunes y que culminan con la activación de un grupo de cisteínas proteasas llamadas “caspasas”, las que llevan a cabo la degradación celular hasta formar pequeños corpúsculos (cuerpos apoptóticos) que son fagocitados por otras células (25). En las células humanas se describen 2 distintas vías apoptóticas:
- 1.
La vía de receptores de muerte oextrínseca, se activa en la membrana plasmática tras la unión de miembros de la superfamilia de receptores de factor de necrosis tumoral (TNF);
- 2.
La vía mitocondrial ointrínseca, se activa por noxas internas y es regulada por proteínas de la familia Bcl-2, conformada por proteínas que inhiben la apoptosis (anti-apoptóticas) y otras que la promueven (pro-apotóticas), y su balance permite la sobrevida o la muerte celular ante estímulos apoptóticos (27).
Las células tumorales desarrollan mecanismos que les permiten evadir la apoptosis. Estos son:
- 1)
Alteración en el balance de proteínas pro- y anti-apoptóticas.
- 2)
Disminución de la actividad de las caspasas.
- 3)
Falla en la señalización del receptor de muerte (28).
Dentro de estas, las mutaciones en las proteínas de la familia Bcl-2 han sido las más estudiadas tras la descripción de la mutación de la proteina Bcl-2 (anti-apoptótica) en el linfoma B no Hodgkin, estirpe B, de tipo folicular, que induce su sobreexpresión, aumentando la sobrevida de las células y por ende, su cantida (29). El desarrollo de drogas destinadas a inhibir la función y expresión de Bcl-2 (para tumores que la sobreexpresan como linfoma y cáncer de próstata), y otros miembros de esta familia podrían restituir las vías de señalización alterada hacia la normalidad, ocasionando la muerte de la célula tumoral que depende de estas fallas para su sobrevida(Figura 2) (28, 30). Estos tratamientos además podrían complementar la eficacia de la radio y quimioterapia cuya acción antitumoral es la inducción de apoptosis, y la integridad de las vías apoptóticas en estas células es fundamental para su eficacia (30).
4) Invasión y metástasis
La mayor parte de las muertes asociadas a tumores sólidos se deben a la diseminación a distancia de las células tumorales y el consecuente desarrollo de metástasis. Del tumor primario, solo algunas células adquieren mutaciones que les permite desarrollar el potencial de invadir el tejido que las rodea (invasión) y posteriormente sitios distantes (metástasis). Dentro de estas mutaciones, la mejor caracterizada es la que provoca la disminución de la expresión de E-cadherina en carcinomas, una proteína fundamental en la adhesividad con otras células y con la matriz extracelular (MEC) por lo que las células tumorales pierden el anclaje al epitelio.
Las células tumorales con capacidad invasiva cambian su fenotipo haciéndose más parecidas a las células mesenquimáticas, en un proceso denominado transición epitelio-mesenquimática (TEM). Las células pierden las uniones adherentes, cambian su morfología epitelial pareciéndose a los fibroblastos, aumentan la expresión de enzimas que degradan la MEC y aumentan su motilidad, lo que favorece la invasión. Por otra parte, los fibroblastos que rodean a las células tumorales también se modifican favoreciendo el proceso carcinogénico y TEM, generando un microambiente pro-tumoral, denominado “estroma reactivo” (15, 31).
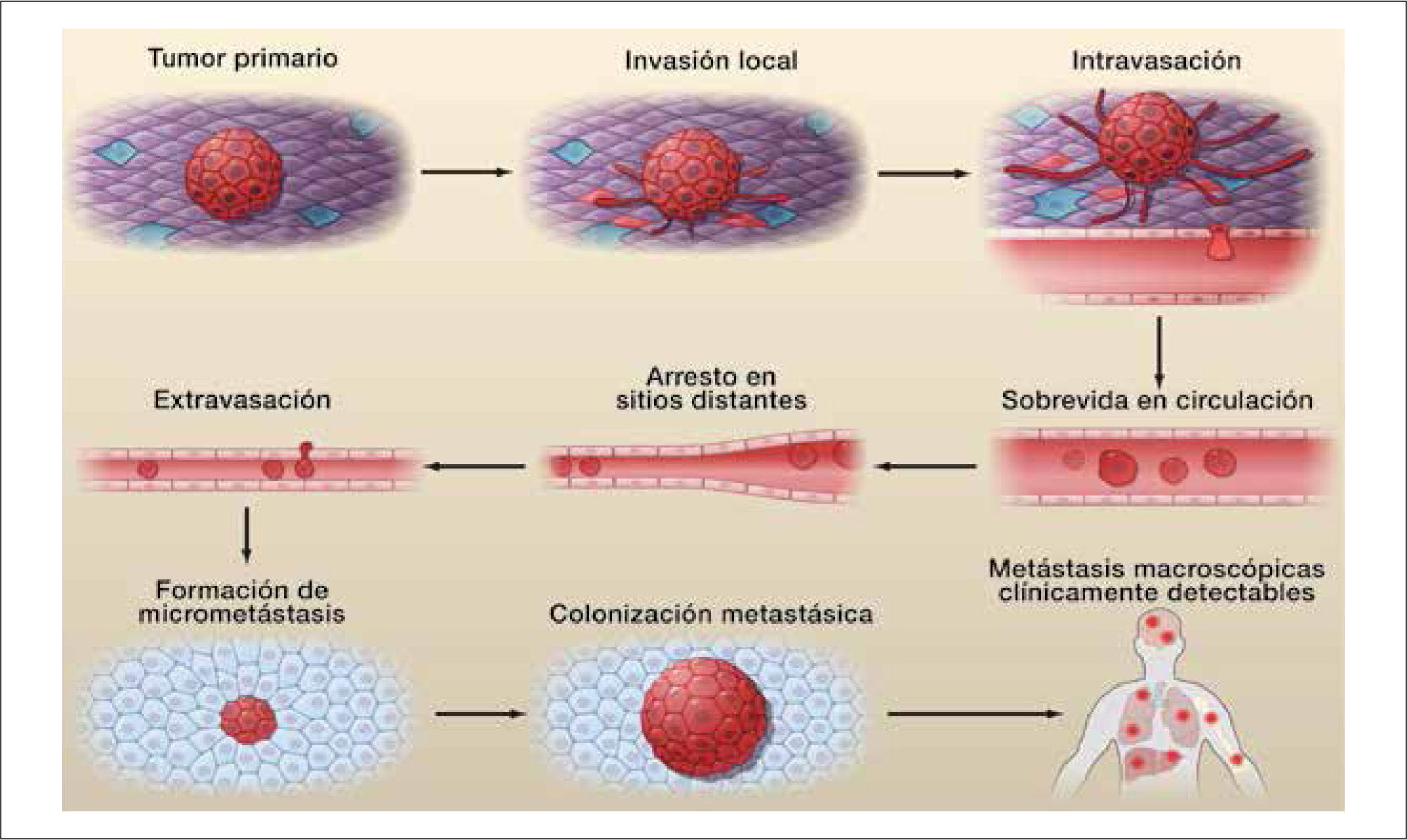
El establecimiento de una metastasis ocurre en una serie de eventos consecutivos denominados cascada invasión-metástasis (Esquema1). A partir del tumor primario, las células comienzan la invasión local, continuando con la intravasación en vasos sanguíneos o linfáticos y tránsito a través de estas vías, la salida de las células desde estos vasos y entrada al parénquima de tejidos distantes (extravasación), concluyendo con la formación de pequeños nódulos de células cancerosas (micrometástasis) que crecen hasta desarrollar tumores macroscópicos (colonización) clínicamente detectables (15).

El proceso invasivo que lleva al desarrollo de una metástasis “exitosa” es un proceso poco eficiente. En cáncer de mama sólo el 0,01% de las células que salen efectivamente a circulación (células tumorales circulantes, CTCs) podría producir una metástasis ósea, y al menos 10.000 CTCs se requieren para el desarrollo de una metástasis exitosa. La presencia de CTCs es necesaria, pero no suficiente para el desarrollo de metástasis, ya que se han encontrado CTCs en pacientes sin enfermedad hasta 20 años después del tratamiento exitoso (31). En los últimos años se ha propuesto la utilidad de la medición de las CTCs y su caracterización funcional con fin pronóstico y predictivo en pacientes afectados por cáncer, aunque aún deben mejorarse los métodos de detección disponibles para su introducción masiva (32).
5) Activación de un potencial de replicación inmortalLa mayor parte de las células pueden pasar por un número limitado de divisiones celulares, lo que se ha denominado límite deHayflick. Cuando las células alcanzan este punto, pueden entrar en dos estados no-proliferativos: la senescencia y la muerte por apoptosis (crisis). En los extremos de los cromosomas hay secuencias múltiples en tandem, denominados telómeros, que se acortan progresivamente en cada división celular hasta un punto que amenaza la estabilidad celular gatillando la crisis celular. Solo algunas células del organismo (como las células germinales reproductivas) pueden sobrepasar este límite y dividirse de manera indefinida, al igual que las células tumorales en un proceso denominado Inmortalización. En estas células se expresa la proteína telomerasa, una ADN polimerasa que adiciona segmentos repetitivos a los extremos del ADN telómerico, evitando su acortamiento. Esta proteína esta ausente en las mayoría de las células del organismo, pero se expresa de manera funcional en cerca de un 90% de las células tumorales espontáneamente inmortalizadas. La expresión de esta proteína junto a mutaciones que inactivan la función de p53, evitan la crisis celular y la muerte o senescencia de las células tumorales, por lo que actualmente se desarrollan drogas que puedan inhibir su función (15).
6) Angiogénesis sostenidaDurante el desarrollo, la formación de nuevos vasos sanguíneos a partir de vasos pre-existentes o angiogénesis es necesaria para suplir de nutrientes y oxígeno a los tejidos en formación. En el cáncer, la estimulación de la angiogénesis favorece el crecimiento tumoral y la metástasis.
La angiogénesis está regulada por diversas vías en las que participan moléculas anti- y pro-angiogénicas que en tejidos normales se encuentran en balance. La pérdida de este balance en el tejido tumoral favorece el desarrollo de nuevos vasos sanguíneos a partir de células precursoras endoteliales (33). El crecimiento tumoral genera hipoxia local que funciona como estímulo para la producción de citoquinas angiogénicas como VEGF, factor de crecimiento de fibroblastos 2 (FGF-2) y factor de necrosis tumoral alfa (TNF-α) entre otras (34).
La variedad de vías moleculares asociadas a la angiogénesis ha abierto una serie de blancos para la intervención terapéutica, principalmente enfocada en la familia VEGF (33). La estimulación de sus receptores VEGF-R1 y 2 activa una vía tirosina quinasa que culmina en vasodilatación, aumento de la permeabilidad vascular, mitosis de células endoteliales y migración. El desarrollo del anticuerpo monoclonal, bevacizumab, que se une a VEGF-A neutralizándolo, ha sido uno de los principales avances terapéuticos en este ámbito, siendo el primero en su clase aprobado por la FDA (34).
7) Reprogramación del metabolismo energéticoEn condiciones normales aeróbicas, las células procesan glucosa hasta su degradación completa a dióxido de carbono en la mitocondria. En células tumorales, ciertas mutaciones y la presencia de un medio pobre en oxígeno conducen al efecto Warburg, una reprogramación del metabolismo energético hacia la glicólisis a pesar de tener oxígeno disponible. La sobreexpresión de transportadores de glucosa (principalmente GLUT1) aumentan su disponibilidad dentro de la célu (15). Se hipotetiza que los tumores requieren una modificación en su metabolismo para cumplir las demandas bioenergéticas y biosintéticas del rápido crecimiento. Además, se propone que este status protegería a la célula de los estados fluctuantes de hipoxia-normoxia que derivan del crecimiento y la re-organización de la nueva vasculatura tumoral (15, 35).
Existe un interés creciente en el desarrollo de agentes terapéuticos para inhibir el efecto Warburg en tumores, sin embargo el riesgo de toxicidad es alto debido a que la glicolisis es un proceso que ocurre en casi todos los tejidos humanos (35).
La alteración del metabolismo celular ha permitido el desarrollo de técnicas de imaginología diagnóstica no invasiva del cáncer. Tal es el caso de la tomografía de emisión de positrones (PET-CT), que utiliza análogos radiomarcados, como 18F-fluorodeoxiglucosa, que se acumula en los tumores ávidos por glucosa y la espectroscopia por resonancia magnética que permite analizar la presencia de metabolitos específicos en tumores (15, 35).
8) Evasión de la destrucción inmuneCuando se desarrollan clones tumorales, estos son normalmente detectados y eliminados por mecanismos inmunológicos (vigilancia inmunológica) que incluyen el sistema inmune innato (monocitos, macrófagos, células natural killers (NK), etc.) y adaptativo (inducción de linfocitos T y B). Fallas en la inmuno vigilancia tumoral podría explicar la mayor incidencia de ciertos tipos de tumores en individuos inmunocomprometidos (36, 37).
Las células tumorales son reconocidas por las células inmunes dado que expresan un patrón proteíco distinto al expresado por células normales, debido a mutaciones, infecciones por virus oncogénicos o por su desregulación y expresión aberrante. Estas proteínas pueden actuar como ligandos que activan células del sistema inmune innato o como antígenos, activando a las células del sistema inmune adaptativo (20). El reconocimiento de las células tumorales no es sencillo, ya que por ser células del mismo individuo, existe cierto grado de inmuno tolerancia. Además, debido a la inestabilidad genómica, las células tumorales cambian constantemente su perfil antigénico, lo que también se acompaña con la sobreexpresión de ciertas proteínas (citoquinas y quimioquinas) que actúan como inmunomoduladores regulando su microambiente y favoreciendo el reclutamiento de monocitos, macrófagos y células inflamatorias altamente supresoras (linfocitos T reguladores y células supresoras derivadas de la línea mieloide (MDSCs)); suprimiendo la actividad del sistema inmune y regulando la neovascularización (15, 36).
AAlgunas células tumorales logran escapar de la eliminación y entran a una fase de equilibrio, en las que son mantenidas en una dormancia funcional de duración indefinida por el sistema inmune adaptativo, hasta que producto de la constante presión de selección, o fallas en la inmunidad, emergen clones que no son reconocidos, escapan del control y continúan proliferando, proceso conocido como inmunoedición (20).
La presencia de células del sistema inmune adaptativo dentro del tumor en algunos tipos de cáncer es un indicador pronóstico. En tumores de colon y ovario, la presencia de linfocitos T citotóxicos y células NK tienen mejor pronóstico que aquellos sin la presencia de linfocitos citotóxicos (15).
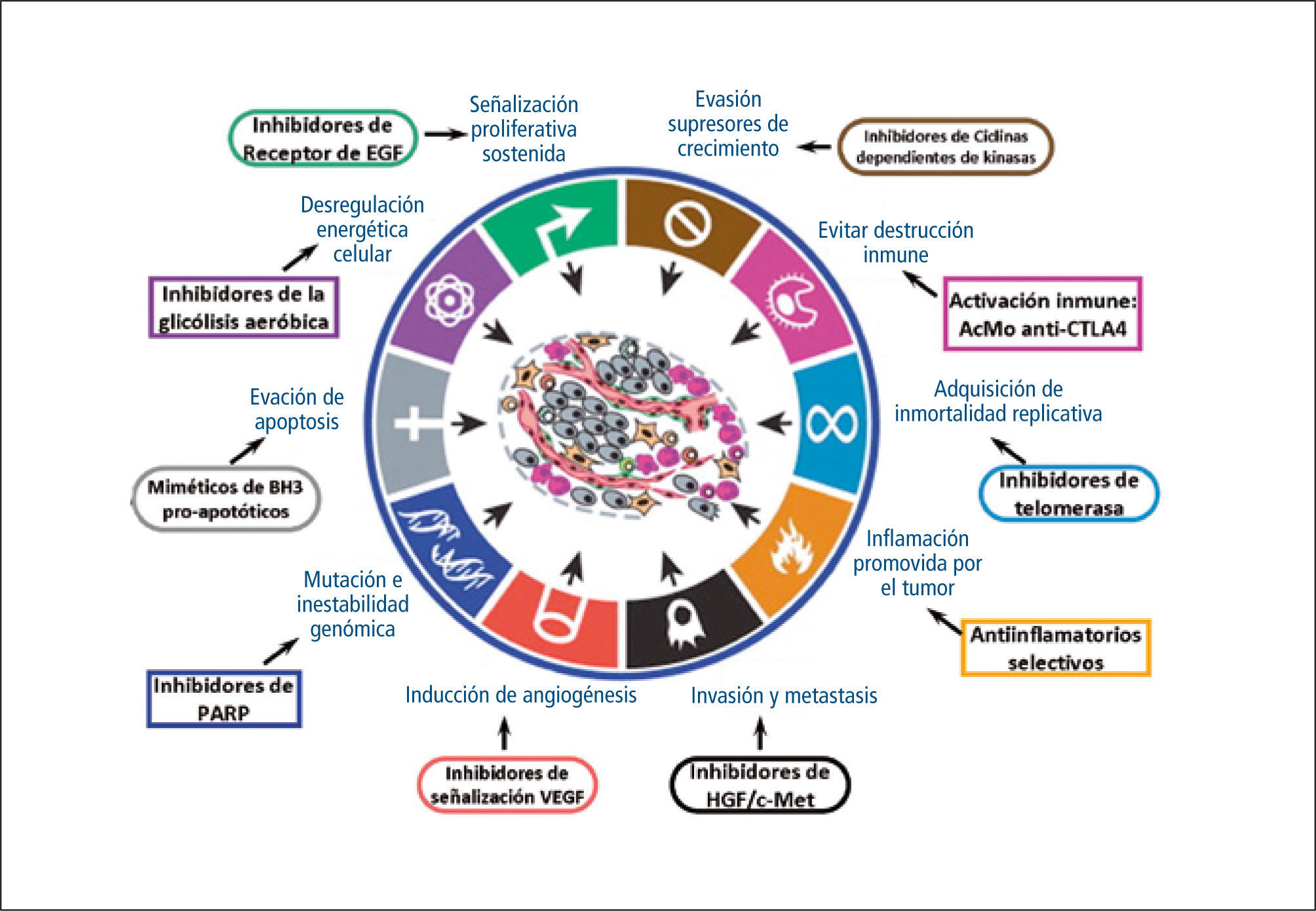
Donde estamos ahora …La revisión presentada aborda parte del conocimiento generado en las últimas décadas sobre la fisiopatología del cáncer, la que ha abierto un abanico de posibilidades para mejorar el manejo del cáncer. Por una parte, la identificación de moléculas que son expresadas sólo por células tumorales ha permitido la introducción de nuevos biomarcadores específicos para mejorar, por ejemplo, el diagnóstico tumoral (como el RNA mensajero PCA3 en cáncer de próstata) o de respuesta a terapia (como la alfa-feto proteína en tumores de células germinales). Por otra parte, la identificación de potenciales blancos terapéuticos ha motivado el incremento explosivo de la investigación farmacológica de la última década. En la figura 3, se mencionan algunas de las terapias en uso actualmente y cómo estas se enfocan a tratar una característica específica que puede verse sobreexpresada en algún tipo tumoral.

El desarrollo e introducción de terapias dirigidas ha logrado mejorar la efectividad antitumoral de las terapias y disminuir los efectos secundarios respecto de las terapias tradicionales. Sin embargo, su implementación hace necesaria la identificación de los mejores candidatos que se verán beneficiados por una terapia en particular. Por esto, se han hecho cada vez más comunes los estudios moleculares, enfocados en la detección de mutaciones específicas o patrones de expresión proteica en los tumores. Existen varios ejemplos de uso rutinario en la clínica, como la detección de la sobreexpresión de erbB2 en cáncer de mama, que es indicador de mal pronóstico y resistencia a tamoxifeno, aunque en otro sentido, es marcador de tumores que responden a trastuzumab. Otro ejemplo es la determinación de mutaciones en el gen KRAS en pacientes con cáncer colorrectal metastático, ya que sólo los pacientes con la forma no mutada (wild) de este gen responden a cetuximab.
La gran cantidad de mutaciones que acumulan las células tumorales, que son adquiridas por distintos mecanismos en los diferentes tipos de cáncer, les confieren ciertas ventajas que finalmente son comunes. Esto permite que muchas terapias sean de uso común en distintos cánceres. Sin embargo, dado el número de mutaciones y la dificultad para encontrar drogas efectivas, en la actualidad la medicina personalizada se ha enfocado en identificar las mutaciones que confieren resistencia a las terapias y a disminuir la toxicidad al paciente debido al uso de terapias inefectivas (12, 38).
Los avances alcanzados en la clínica van muchas veces de la mano con los avances tecnológicos. La introducción de técnicas genómicas de alto rendimiento (high-throughput genomic) permite realizar perfiles a los tumores, facilitando la búsqueda de nuevos blancos diagnósticos y terapéuticos. Por otra parte, ha crecido el desarrollo de perfiles genéticos únicos o múltiples que tienen variadas aplicaciones: en el establecimiento del riesgo de padecer cáncer, en el diagnóstico, pronóstico y terapia. Su uso como test genético a familias afectadas por algunos tipos de tumores (en particular colon, pulmón, mama), ha permitido mejorar significativamente el manejo de los pacientes y sus parientes (10). Durante los próximos años se espera incrementar estas herramientas gracias a la creciente investigación en microRNAs y el establecimiento de perfiles de expresión específico para los distintos tipos tumorales, lo que abrirá a futuro una nueva área de crecimiento en el área de diagnóstico y terapia.
Los frutos generados por la investigación médica básica deben ser complementados por la investigación clínica y epidemiológica; sólo esto permitirá optimizar recursos y obtener mejores resultados para el paciente. El desarrollo de la medicina traslacional depende del entendimiento entre estos tres ámbitos de investigación, su confluencia e interacción. La participación de médicos en la investigación básica es cada vez más fundamental para acelerar su desarrollo, mejorar su calidad y el alcance y aplicabilidad de los resultados obtenidos.
La autora declara no tener conflictos de interés, con relación a este artículo.