



La fenitoína es usada con mucha frecuencia en nuestro medio, por lo que se requiere hacer estudios de monitorización terapéutica, que contribuya a minimizar los efectos adversos y optimizar la terapia farmacológica. En ese contexto, nuestro objetivo ha sido determinar el índice nivel/dosis de la fenitoína en pacientes epilépticos voluntarios de Mérida.
MétodosSe realizó un estudio descriptivo, observacional y por reclutamiento consecutivo concurrente, conformado por 30 pacientes voluntarios con diagnóstico de epilepsia. Las muestras de suero se obtuvieron en niveles mínimos de pacientes que estaban en tratamiento con fenitoína durante 1 mes. Los niveles del fármaco se cuantificaron por el método de Inmunoensayo de enzima donante clonada en el equipo Indiko Thermo Scientific.
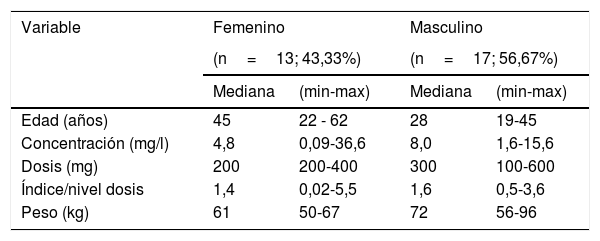
ResultadosEl índice nivel/dosis fue de 1,4 y 1,6, la concentración plasmática de 4,8mg/l y 8,0mg/l, la capacidad metabólica de 388,4 y 462,9mg/día, respectivamente en mujeres y hombres. Mientras que el nivel de la concentración plasmática en el estado estacionario fue de 6,5mg/l y 5,5mg/l, la dosis de carga máxima de 237,3mg y de 395,6mg, respectivamente en mujeres y hombres con epilepsia de la ciudad de Mérida.
ConclusionesNuestros resultados sugieren que se debe individualizar la dosis en base al índice nivel/dosis de cada paciente, ya que no se puede extrapolar para todos los pacientes con epilepsia, debido a diversos factores como al fenotipo metabólico y al uso de fármacos inductores e inhibidores enzimáticos.
Phenytoin is used very frequently in our environment, so it is necessary to do studies of therapeutic monitoring, which helps to minimize adverse drug reaction and optimize pharmacological therapy. In this context, our objective was to determine the level/dose index of phenytoin in volunteer epileptic patients from Mérida.
MethodsA descriptive, observational and consecutive concurrent recruitment study was carried out, consisting of 30 volunteer patients with a diagnosis of epilepsy. The serum samples were obtained in minimum levels from patients who were in treatment with phenytoin for 1 month. The levels of the drug were quantified by the method of donor enzyme immunoassay cloned in the Indiko Thermo Scientific equipment.
ResultsThe level/dose index was 1,4 and 1,6, the plasma concentration of 4,8mg/l and 8,0mg/l, the metabolic capacity of 388,4 and 462,9mg/day, respectively in women and men. While the level of plasma concentration at steady state was 6,5mg/l and 5,5mg/l, the maximum loading dose of 237,3mg and 395,6mg, respectively in women and men with epilepsy of the city of Mérida.
ConclusionsOur results suggest that the dose should be individualized based on the level/dose index of each patient, since it can not be extrapolated for all patients with epilepsy, due to various factors such as the metabolic phenotype and the use of enzyme-inducing drugs and inhibitors.
La International League Against Epilepsy (ILAE) define a la epilepsia como una enfermedad neurológica con la presencia de dos o más crisis convulsivas, que se presentan en más de 24 horas por separado o una crisis convulsiva no provocada con una alta probabilidad de al menos 60% de recurrencia dentro de los siguientes 10 años1–3. Se ha reportado que en el mundo unos 50 millones de personas tienen epilepsia, y casi el 80% de los casos se presentan en los países en vías de desarrollo, en donde se utilizan como tratamiento estándar a los anticonvulsivantes4. Las concentraciones plasmáticas de estos fármacos deben ser monitorizados a través de la farmacocinética clínica, y mediante la farmacogenética se debe estudiar el genotipo del paciente para determinar el fenotipo metabólico. La Medicina de Precisión se basa en ambas ciencias para adaptar el tratamiento a cada paciente, con la dosis correcta y en el momento correcto, cuyo objetivo es minimizar los efectos adversos y optimizar la terapia farmacológica5–8.
La fenitoína (PHT), químicamente denominada 5,5-difenilimidazolidina-2,4-diona, es de naturaleza ácida, con un pKa de 9,2, soluble en agua (14mg/l), un factor de sal sódica de 0,92, y una cinética de absorción saturable no lineal de orden cero (50mg/h). Su biodisponibilidad es de 80%9, la cual se reduce por interacciones farmacológicas10 o por nutrición enteral, en el síndrome de malabsorción o por infecciones gastrointestinales11. Este fármaco es de estrecho margen terapéutico (MT), debido a que su concentración mínima efectiva (CmE) es de 10mg/l y su concentración máxima efectiva (CME) es de 20mg/l120,13. Su índice terapéutico es de 2mg/l, el cual se obtiene al hacer la relación CME/CMe.14 Por lo tanto, niveles por debajo de la concentración mínima aumenta el riesgo de convulsiones; mientras que los niveles superiores a la concentración máxima predisponen a la generación de toxicidad13,15. Su tiempo máximo es de 1-6 horas en comprimidos de liberación inmediata y de 4-12 horas en sistemas de liberación prolongada9. El estado de equilibrio estacionario (Css) se alcanza entre 3 a 50 días10. El volumen aparente de distribución (Vd) es de 0,65l/kg a 0,8l/kg11; en caso de insuficiencia renal se incrementa a 1,4l/kg. El rango de semivida (t1/2) fluctúa entre 30-100 horas9 con un promedio de 6-12 h10 con dosis baja y de 12-60h con una dosis alta10,11. La fenitoína se une a las proteínas plasmáticas (UP) en un 90%, y esta fracción no atraviesa la barrera hematoencefálica (BHE) 11,16. Se puede presentar un desplazamiento de su UP por el uso conjunto de AINEs, ácido valproico, heparina, sulfonamidas y warfarina. En caso de hipoalbuminemia e insuficiencia hepática, la fenitoína queda libre sin unirse a la proteína y sin metabolizarse, lo que podría generar reacciones adversas al superar la concentración plasmática máxima. Por esta razón, es recomendable la medición de niveles plasmáticos de fenitoína libre (1-2mg/l) en lugar de los niveles totales11. La PHT se metaboliza en 2 fases, primero con la participación del CYP2C9 (90%)16–18 y CYP2C1919 generando el metabolito inactivo p-hidroxifenitoína (p-HPPH). Luego se conjuga con el ácido glucurónico20, por acción de la enzima uridina-5-difosfo glucuronosiltransferasa 1A (UGT1A) formando al O-β-glucurónido de fenitoína el cual se elimina por la orina; un menor porcentaje del p-HPPH se oxida y forma catecol fenitoína, con participación del CYP2C19 y CYP2C920. Su metabolismo hepático es mayor al 95% de acuerdo a una cinética de orden cero. La capacidad metabólica (Vm) de un adulto es de 6 a 7mg/kg/día y su constante metabólica (Km) es de 4mg/l. Se ha demostrado que los fármacos inductores del metabolismo de la fenitoína (carbamazepina, clonazepam, diazepam, clordiazepóxido, dexametasona, fenobarbital, vigabatrina, alcohol, ácido fólico, nitrofurantoína y rifampicina) aumentan la capacidad metabólica; mientras que los fármacos inhibidores (AINEs, ácido valproico, felbamato, progabida, amiodarona, anticoagulantes orales, alopurinol, cimetidina, clorpromazina, metronidazol, ranitidina, sulfonamidas, imipramina, omeprazol, isoniazida) aumentan la constante de metabolización. La fenitoína es inductor de su propio metabolismo y es inductor enzimático de diversos fármacos disminuyendo su concentración plasmática y su semivida del topiramato21, perampanel22, carbamazepina, fenobarbital, tracolimus, warfarina y tamoxifeno23.
Las variantes alélicas CYP2C9*2 y CYP2C9*3 son predictores del fenotipo metabólico lento8,18,19. El CYP2C9*2 se caracteriza por una transición citosina por timina en el par de bases 430 del exón 3 (430C>T, rs 1799853), que conduce a la sustitución de arginina por cisteína en el codón 144 (Arg144Cys) con una actividad aproximada del 12% respecto al wild type debido a una menor interacción con el cofactor8,24; y el CYP2C9*3 se produce por una transversión de adenina por citosina en el par de bases 1075 del exón 7 (1075A>C, rs 1057910) que da como resultado un cambio de isoleucina a leucina en el codón 359 (Ile359Leu) con una actividad del 5% debido a una variación en el sitio de unión al sustrato24. Dichos genes, condicionan sus parámetros farmacocinéticos por disminución en su metabolismo20. Se ha reportado en diversos estudios que los genotipos CYP2C9*1/*2, CYP2C9*1/*3,25 CYP2C9*2/*2 y CYP2C19*1/*4, están implicados en los efectos adversos de la fenitoína en caucásicos18; en los indios es el CYP2C9*3/*326, en japoneses es el CYP2C9*1/*3 y CYP2C19*1/*3, y en afroamericanos están implicados los genes CYP2C9*6/*6 y CYP2C19*1/*127. Desde el punto de vista de la farmacocinética, se tiene evidencia que al superar los 20mg/l algunos pacientes presentan somnolencia, fatiga, nistagmo y diplopía; por sobre los 30mg/l se observa ataxia e incoordinación; y a concentraciones mayores de 40mg/l, se observa confusión, letargia y coma; un efecto adverso característico es la hiperplasia gingival, hirsutismo, dermatitis alérgica, piel áspera, acné, anemia aplásica, anemia megaloblástica, agranulocitosis, hepatotoxicidad, pancreatitis y teratogenicidad27.
El índice nivel/dosis (N/D) es un método que nos permite valorar cuantitativamente el nivel sérico hipotético alcanzado con una dosis teórica de 1mg/kg y se utiliza para detectar el incumplimiento en los fármacos con cinética lineal, nos permite ajustar la dosis con precisión y puede ser de utilidad clínica para realizar la medicina de precisión5,28. El N/D varía entre distintos individuos y poblaciones, en función de la edad, del consumo de diversos fármacos e incorrecto cumplimiento de la prescripción; en monoterapia es mayor que en la politerapia, por la interacción con otros antiepilépticos inductores enzimáticos que aumentan la eliminación del antiepiléptico28.
En Latinoamérica los estudios sobre el índice nivel/dosis y sobre la capacidad metabólica en pacientes epilépticos son muy limitados, en Chile, Escobar L., reporta que, entre los anticonvulsivantes, la fenitoína es uno de los fármacos que se monitoriza en suero y sus concentraciones valle deben ser de 10-20mg/l y no se requiere una concentración máxima plasmática;12 Guevara et al., en Uruguay, observaron en 50 pacientes epilépticos caucásicos, que los genotipos CYP2C9 y CYP2C19 influyen sobre las concentraciones plasmática de la fenitoína y del p-HPPH20.
Luego de realizar una revisión en la base de datos del PubMed-NCBI sobre estudios de nivel dosis (N/D) y capacidad metabólica, se ha evidenciado la ausencia de investigaciones en pacientes con epilepsia en Venezuela. Estos estudios son necesarios, ya que al conocer los niveles plasmáticos de los fármacos de estrecho margen terapéutico, nos permiten proponer dosis con mayor precisión. En tal sentido, hemos decidido estudiar a la fenitoína por su índice terapéutico, por su metabolismo saturable y por su alta variabilidad farmacocinética, lo que hace imprescindible una monitorización terapéutica continua, para prevenir su toxicidad y asegurar su eficacia29,30.
El objetivo del presente estudio fue determinar el índice nivel dosis de la fenitoína en pacientes epilépticos voluntarios de Mérida.
MétodosDiseño y población de estudioSe realizó un estudio observacional, descriptivo y transversal, ya que la investigación describió, midió y distribuyó el fenómeno dentro de la población estudiada; y por reclutamiento consecutivo concurrente, al ser seleccionados todos los pacientes que acudían consecutivamente para su control y tratamiento. La población de estudio fueron pacientes con diagnóstico de epilepsia que acudían al consultorio externo del Instituto Autónomo Hospital Universitario de Los Andes, Mérida, Venezuela, entre enero de 2017 a diciembre de 2018; cuyas edades comprendían de 19 a 62 años con una mediana de 45 años en las mujeres y de 28 años en los hombres, los mismos, que luego de firmar el consentimiento informado se les denomino pacientes voluntarios.
Variables y medicionesLa variable dependiente son los niveles de fenitoína, como definición operacional los niveles plasmáticos entre 10-20mg/l y una dimensión de concentración máxima tolerada y una mínima inhibitoria. Entre las variables independientes se consideran el sexo, edad, peso y nacionalidad.
Análisis estadísticoEl análisis estadístico se realizó utilizando el programa Graphad Prism versión 5.00.
Consideraciones éticasEl estudio fue desarrollado en estricto cumplimiento de las normas éticas internacionales y en base al consentimiento informado aprobado mediante constancia N°001-17-UN-FM-ULA. Los pacientes fueron seleccionados de acuerdo a los siguientes criterios de inclusión: pacientes en tratamiento con fenitoína con no menos de un mes de terapia farmacológica, que no consuman otros medicamentos sin prescripción de su médico tratante, que al examen médico no manifieste daño renal ni hepático y que antes de las 24 horas de la toma de la muestra no consuma bebidas y alimentos que contenga cafeína u otro metabolito que pueda interaccionar con la fenitoína. Antes de la toma de la muestra se interrogó a cada paciente sobre los criterios cumplidos y de ayuno, siendo excluidos todos aquellos que no cumplieron con dichos criterios5.
Técnica y metodología de estudioSe obtuvo una sola muestra de sangre de cada uno de los 30 pacientes voluntarios con diagnóstico de convulsiones y que recibían fenitoína sódica tres veces al día como dosis estándar. Las muestras de sangre se colectaron como parte de la rutina del tratamiento y del seguimiento farmacoterapéutico. Todas las muestras sanguíneas se colectaron en tubos BD VacutainerR de 3ml, la misma que se realizó después de 30 días de tratamiento, para asegurar que las concentraciones estén en el estado estacionario, y en condiciones de ayuno (entre las 8:00-10:00 a.m.) y con no menos de 10 horas después de la última toma de la dosis y antes de la administración del medicamento del presente día5,12. Para la cuantificación de la fenitoína en plasma, primero se centrifugó 2ml de sangre de cada paciente a 8000g por 10min, luego se midió 500 microlitros de suero y se analizaron por duplicado por el método de CEDIA (Inmunoensayo de enzima donante clonada) en el equipo Indiko Thermo Scientific.5 Los análisis de cuantificación fueron realizados por los Farmacéuticos Toxicólogos del Departamento de Farmacia de la Universidad de Los Andes de Mérida.
El ND se calculó al dividir la concentración plasmática alcanzada en el estado estacionario (Css) entre la dosis/kg [ND=Css/(Dmg/kg)]5,28. El nivel de la concentración en el estado estacionario (Css) se determinó mediante la siguiente ecuación: Css=(Dosis única/Peso kg) x 4. La dosis de carga (DQ) se determinó con la ecuación: DQ=Vd x CmE/S x F, donde S es el factor de la sal de la fenitoína que corresponde a 0,92 y F (biodisponibilidad). La capacidad metabólica (Vm) se determinó con la ecuación: Vm=(S)(F)(Dosis/τ)/(Km+Css promedio); y el clearance con la ecuación: Cl=Dosis/τ (intervalo de administración.
ResultadosSolo los pacientes que cumplieron con los criterios de inclusión del Servicio de Neurología del Instituto Autónomo Hospital Universitario de Los Andes de Mérida, fueron incluidos en el estudio. Todos fueron mayores de edad, de ambos sexos, en tratamiento con fenitoína sódica a una dosis estándar que era dividida en tres dosis, con una mediana de N/D de 1,4 para las mujeres y de 1,6 para los hombres (Tabla 1).
Descripción de variables clínicas de los pacientes voluntarios de Mérida
| Variable | Femenino | Masculino | ||
|---|---|---|---|---|
| (n=13; 43,33%) | (n=17; 56,67%) | |||
| Mediana | (min-max) | Mediana | (min-max) | |
| Edad (años) | 45 | 22 - 62 | 28 | 19-45 |
| Concentración (mg/l) | 4,8 | 0,09-36,6 | 8,0 | 1,6-15,6 |
| Dosis (mg) | 200 | 200-400 | 300 | 100-600 |
| Índice/nivel dosis | 1,4 | 0,02-5,5 | 1,6 | 0,5-3,6 |
| Peso (kg) | 61 | 50-67 | 72 | 56-96 |
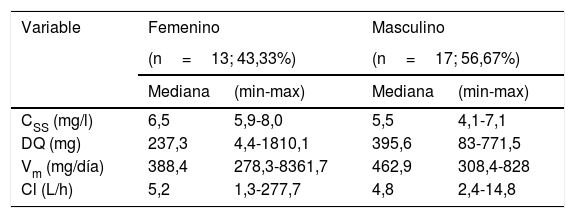
En la tabla 2 se reporta los parámetros farmacocinéticos como el nivel de la concentración plasmática en el estado estacionario (Css), la dosis de carga máxima (DQ), la capacidad metabólica (Vm) y el clearance (Cl) para los pacientes con epilepsia de la ciudad de Mérida.
Parámetros farmacocinéticos obtenidos para pacientes con diagnóstico de epilepsia de Mérida
| Variable | Femenino | Masculino | ||
|---|---|---|---|---|
| (n=13; 43,33%) | (n=17; 56,67%) | |||
| Mediana | (min-max) | Mediana | (min-max) | |
| CSS (mg/l) | 6,5 | 5,9-8,0 | 5,5 | 4,1-7,1 |
| DQ (mg) | 237,3 | 4,4-1810,1 | 395,6 | 83-771,5 |
| Vm (mg/día) | 388,4 | 278,3-8361,7 | 462,9 | 308,4-828 |
| Cl (L/h) | 5,2 | 1,3-277,7 | 4,8 | 2,4-14,8 |
En nuestro estudio se utilizó la técnica de inmunoensayo de enzima donante clonada, la misma que es específica para cuantificar fenitoína, con pocas interferencias reportadas. Una de las posibles interferencias en las mediciones de fenitoína es la reactividad cruzada que se genera entre el metabolito 5-(p-hidroxifenil)-5-fenilhidantoína y su metabolito glucurónido de fenitoína que se excreta en la orina. Esta reactividad es importante en pacientes con insuficiencia renal debido al aumento del metabolito conjugado por su dificultad de eliminación, y que no ha sido en nuestro caso31. El estudio describió las variables clínicas y la distribución de los pacientes voluntarios por género, observándose que la mediana de las concentraciones plasmáticas es de 4,8mg/l y 8,0mg/l, en mujeres y hombres, respectivamente, lo que se encuentra por debajo del valor internacional de 10-20mg/l, de acuerdo a lo reportado por Escobar L,12 esto a pesar de haber tomado las muestras de sangre después de los 30 días de iniciado el tratamiento, cuya finalidad fue, asegurar un nivel plasmático en el estado estacionario. Uno de los factores que podrían influir en este parámetro es la variabilidad de la semivida y la interacción del fármaco con los nutrientes.
El N/D encontrado en nuestro estudio es de 1,4 y 1,6, la capacidad metabólica de 388,4mg/día y 462,9mg/día, en mujeres y hombres respectivamente. Sabemos que el metabolismo de la fenitoína sigue una cinética lineal a bajas concentraciones, pero al saturarse sigue una cinética de orden cero. Esto es crítico en la clínica, ya que una variación mínima de la dosis, una variante alélica de los genes CYP2C924 y CYP2C1927 responsables de su metabolismo, el uso de fármacos inhibidores enzimáticos o el desplazamiento de su unión a proteínas, pueden generar un incremento del N/D y, por lo tanto, generar efectos adversos o toxicidad, tal como lo reporta Hernández L et al.,3 y Guevara N et al.20
Nuestro valor, se encuentra en el límite inferior propuesto por Sánchez et al., quienes hicieron una revisión sobre el valor del N/D de la fenitoína en monoterapia y que a partir de los 15 años de edad es de 2-3; mencionando que es útil para el ajuste de dosis y para detectar el incumplimiento en fármacos con cinética lineal, y que varía entre distintos individuos y por la politerapia27; pero ya en el año de 1968 Kutt y McDowell, habían demostrado que los niveles de la fenitoína son variables, las mismas que se deben a una deficiente p-hidroxilación del fármacos por disfunción hepática o por la interacción con diferentes fármacos como la isoniazida, clorpromazina, estrógenos, disulfiram y otros32.
Posteriormente Borofsky et al (1972) reportaron que el N/D de la fenitoína varía con la edad, encontrando en 14 de los 53 pacientes un valor de 1-3, siendo mayor en pacientes con edad superior a 16 años33. En un estudio realizado por Lee et al (1981), se observaron que el N/D de la fenitoína era menor a 1, en más del 73% de los 30 niños epilépticos chinos estudiados, ya que se tiene documentado que el N/D en niños debe ser de 1-2, concluyendo que el control deficiente de las convulsiones en dichos pacientes se debió principalmente a concentraciones subterapéuticas de fenitoína, y que no se pueden atribuir al mal cumplimiento del paciente34. Rasheva M et al., manifestaron en su estudio que un bajo nivel sérico de los fármacos antiepilépticos, se debe entre un 20-50% a una falta de adherencia e incumplimiento de la terapia farmacológica; a la vez se reporta, que la interacción farmacocinética es el otro factor del bajo nivel sérico de los anticonvulsivantes35. En otro estudio Panomvana D et al., han establecido que la concentración sérica de la fenitoína varía de 1,40 a 16,28mg/l, la misma que se estableció luego de la administración conjunta con la carbamazepina, que es un inductor enzimático36. Con el N/D encontrado proponemos el nivel de la concentración plasmática en el estado estacionario (Nivel Css 6,5mg/l en mujeres y 5,5mg/l en hombres) y la dosis de carga (DQ 237,3mg en mujeres y 395,6mg en hombres) como dosis inicial para alcanzar el estado estacionario de manera rápida.
Las limitaciones de nuestro estudio están en el tamaño de la muestra (n=30) que no es representativa de la población de estudio, la selección de la misma fue por conveniencia y no aleatoria, no se calculó el tamaño muestral a partir de pacientes con diagnóstico de epilepsia. Otro sesgo, que puede inducir a confusión y que no fueron medidas fueron los años de enfermedad y los años de uso de fenitoína como terapia farmacológica. Pero la importancia de nuestro estudio radica en obtener evidencia científica del N/D que es muy pobre en la fenitoína y valproato, de la capacidad metabólica, del proceso de depuración renal y de otros parámetros farmacocinéticos, en nuestras poblaciones latinas, y que ello nos permita detectar el incumplimiento de la prescripción, detectar la interacción con fármacos inductores enzimáticos y ajustar la dosis con precisión, cuya finalidad es minimizar los efectos adversos y optimizar la terapia farmacológica. Tanto la farmacocinética clínica como la farmacogenética, son las ciencias básicas que dan sustento a la medicina de precisión, la misma que es necesaria en las enfermedades crónicas como la epilepsia, por lo que estamos promoviendo su implementación en nuestro medio.
En conclusión, nuestros resultados sugieren que se debe individualizar la dosis en base al N/D de cada paciente, ya que no se puede extrapolar para todos los pacientes con epilepsia, debido a diversos factores como al fenotipo metabólico, al uso de fármacos inductores e inhibidores enzimáticos y a las limitaciones descritas.
Fuentes de financiamientoLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflictos de interésDeclaración de conflictos de interés
Los autores declaran no tener ningún conflicto de interés.