





Por más de 60 años se dispuso solo de tres grupos farmacológicos para el tratamiento de la diabetes mellitus (DM): la insulina, la metformina y las sulfonilureas. Sin embargo, en los últimos años y como consecuencia de los avances en el conocimiento de la patogenia de la DM2 se han desarrollado nuevos fármacos con novedosos mecanismos de acción y con diferentes perfiles de seguridad, entre ellos los compuestos con efecto incretina y los glucosúricos que actúan en los trastornos a nivel intestinal y renal presentes en la DM2. La disponibilidad de múltiples opciones terapéuticas está produciendo profundos cambios en la terapia farmacológica de la DM2. Se logran enfoques terapéuticos más fisiopatológicos pero sobre todo permiten un manejo más personalizado y ajustado a las características y riesgos individuales de los pacientes, privilegiando junto al control glicémico, la seguridad terapéutica. En esta revisión se analizarán los nuevos fármacos con efecto incretina, los agonistas del péptido similar al glucagón tipo 1 (AR-GLP1) e inhibidores de la dipeptidil peptidasa 4 (IDPP-4), y los inhibidores de los cotransportadores sodio-glucosa tipo 2 (ISLGT2) que aumentan la excreción renal de glucosa.
For over 60 years only three drug classes, insulin, metformin and sulfonylureas, were available for the treatment of Diabetes Mellitus (DM). In recent years, as a result of advances in the understanding of the pathogenesis of type 2 diabetes, new drugs with novel mechanisms of action and different safety profiles have been developed, including compounds with incretin effect and glucosuric drugs acting in disorders at intestinal and renal level present in DM2. The availability of multiple treatment options is leading to profound changes in drug therapy of type 2 diabetes. More pathogenic therapeutic approaches are accomplished, allowing a more personalized and tailored management to the individual characteristics and risks of patients, achieving, in addition to glycemic control, therapeutic safety. In this review, the new class of antihyperglycaemic agents with incretin effect, glucagon-like peptide-1 receptor agonists (GLP-1 RAs) and dipeptidyl peptidase-4 inhibitors (IDPP-4), and novel sodium-glucose cotransporter 2 inhibitors (ISGLT2) that increase renal glucose excretion will be analyzed.
La Diabetes Mellitus (DM) ha llegado a ser uno de los principales problemas de salud debido a su creciente prevalencia, su contribución al desarrollo de patologías vasculares crónicas y a su elevada mortalidad 1,2.
El tratamiento de la DM tiene como objetivos asegurar al paciente una buena calidad de vida, disminuir el riesgo de complicaciones específicas (retinopatía, nefropatía, neuropatía) y de eventos cardiovasculares (CV) que son su principal causa de mortalidad. Hay consenso que, para lograrlo, se requiere un enfrentamiento terapéutico multifactorial con control de la hiperglicemia, y de otros factores de riesgo CV, habitualmente presentes en las personas con DM3. Sin embargo, la piedra angular del manejo de la DM es el control glicémico4. Es la herramienta terapéutica más eficaz para reducir el riesgo de desarrollo y/o progresión de la enfermedad microvascular5, y, si bien el impacto en las complicaciones macrovasculares sigue siendo un tema de debate, un metaanálisis de varios ensayos clínicos prospectivos y controlados muestra que el mejor control metabólico per sé se asocia con la reducción de la incidencia de eventos CV6.
Hasta el año 1920, en que se descubrió la insulina, no se disponía de fármacos para el manejo de la hiperglicemia7. En los años 50 se incorporaron las sulfonilureas (SU) y la metformina (MF), y por casi 50 años estas 3 clases de drogas fueron las únicas disponibles para el tratamiento de la DM. En 1995 se agregan los inhibidores de las alfa glucosidasas, que enlentecen la absorción de glucosa a nivel intestinal. Poco después las tiazolidinedionas (TZD) con su efecto insulinosensibilizador y las meglitinidas con su acción secretora prandial8. Hasta entonces para el tratamiento de la DM2 se contaba con fármacos que actuaban estimulando la secreción de insulina o mejorando su sensibilidad en los tejidos, corrigiendo así lo que se consideraba eran los dos principales mecanismos patogénicos de esta enfermedad: disfunción de la célula β y resistencia a la insulina (RI) en tejido hepático, adiposo y muscular7,8.
En la década del 2000, De Fronzo describe al “octeto ominoso”9, para referirse a los múltiples y complejos mecanismos patogénicos de la DM2. A la RI y falla de secreción β insular se agregan otras alteraciones y órganos involucrados en su patogenia como la disminución del efecto de las incretinas intestinales, el aumento de la producción de glucagón por las células α, la disfunción de neurotransmisores a nivel cerebral y el aumento de la reabsorción de glucosa renal9. Este mayor conocimiento de la fisiopatología de la DM2da origen al desarrollo de nuevos fármacos con novedosos mecanismos de acción. Primero aparecen preparados con efecto incretina que se dividen en agonistas del péptido similar al glucagón tipo 1 (AR-GLP1) introducidos para el uso clínico en el año 2005 y los inhibidores de la dipeptidil peptidasa 4 (IDPP4), disponibles desde el 2006. Recientemente se agregan los inhibidores de los cotransportadores de sodio-glucosa tipo 2 (ISGLT2), que focalizan su acción hipoglicemiante a nivel renal. También se cuenta con fármacos agonistas de dopamina, con mecanismos de acción central aún no bien definidos10 y análogos de amilina, que disminuyen la secreción de glucagón post prandial (pp) y modulan el vaciamiento gástrico (VG). Estos últimos son raramente usados en la práctica clínica y tienen modesto efecto en la hemoglobina glicosilada A1c (HbA1c)10. La creciente disponibilidad de antidiabéticos para el manejo de la DM2 permite en la actualidad variadas opciones para lograr los objetivos glicémicos y elegir la terapia que mejor se ajuste a las características individuales de cada paciente. En esta revisión se analizarán los dos grupos de fármacos con efecto incretina, los agonistas del receptor de GLP-1 e inhibidores de DPP4 y el nuevo grupo de glucosúricos, los inhibidores de SGLT2, con foco en sus mecanismos de acción, efectividad y seguridad terapéutica.
I. FÁRMACOS CON EFECTO INCRETINAEL EFECTO INCRETINALas hormonas incretinas son péptidos liberados en el intestino en respuesta a la presencia de nutrientes en el lumen intestinal11. El efecto incretina, como se denomina a la acción de estas hormonas, forma parte del eje enteroinsular de la homeostasis de la glucosa y se estima que es responsable del 50% al 70% de la secreción de insulina12. Fue descrito al observar que la administración de glucosa oral se asocia con un aumento mucho mayor en los niveles plasmáticos de insulina en comparación con la respuesta secretora pancreática que se obtiene con cantidades equivalentes de glucosa administrada por vía endovenosa13.
Las principales incretinas son el péptido similar al glucagón-1 (GLP-1 glucagon-like peptide 1) y el péptido insulinotrópico dependiente de glucosa (GIP glucose-dependent insulinotropic peptide)11. El GLP-1 es producido por las células enteroendocrinas de las células L del íleon distal y colon y el GIP es elaborado por las células K del duodeno y el yeyuno. Ambas hormonas son rápidamente liberadas después de la ingesta, al parecer bajo control neural, y estimulan la producción de insulina en las células β pancreáticas de una manera dependiente de la glucosa. Además, GLP-1 disminuye la secreción de glucagón desde las células α pancreáticas, retarda el VG y es probable que tenga un efecto supresor directo sobre centros del apetito11. Se ha demostrado que en pacientes con DM2 la respuesta al efecto incretina está alterada14 como resultado de un severo defecto de la sensibilidad para el GIP en la célula Beta y de reducción en la secreción de GLP-1 inducida por alimentos15. Sin embargo, a pesar de que la secreción post prandial de GLP-1 está deteriorada, sus acciones se conservan16. Recientemente se cuestiona si la disregulación incretina es causa o consecuencia de la hiperglicemia de la DM217.
Mientras que GIP actúa casi exclusivamente a nivel β pancreático, existen receptores para GLP-1(R-GLP1) ampliamente distribuidos en el páncreas endocrino (alfa y β), en órganos periféricos (corazón, estómago, nervio vago) y en varias regiones del SNC, incluyendo el hipotálamo y el tronco cerebral. Estudios en animales han demostrado que GLP-1 promueve la neogénesis y la proliferación celular, inhibe la apoptosis e incrementa la masa de células β11. Por otra parte se han descrito una serie de efectos positivos en vías involucradas en la aterogénesis vascular, función endotelial y protección ventricular18. De manera que a esta incretina además de regular el control de la glucosa se le atribuyen probables efectos protectores a nivel β insular, neural y CV.
Las formas biológicamente activas de GIP y GLP-1 tienen una vida media plasmática corta (<2min), al ser rápidamente inactivadas por la acción proteolítica de la enzima dipeptidil peptidasa 4 (DPP-4)19. Esto limitó el entusiasmo inicial de la posible utilidad de las incretinas para el tratamiento de la DM2, y a su vez estimuló el desarrollo de fármacos agonistas del R-GLP1 resistentes a DPP-4, con vida media más larga y de inhibidores de la DPP-4 que prolongan la vida media de las incretinas nativas.
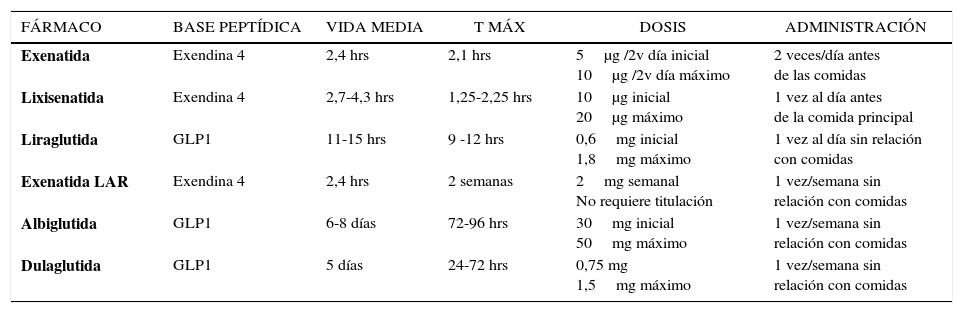
1. AGONISTAS DEL RECEPTOR DE LA GLP1 (AR-GLP1)Los AR-GLP1 son un grupo de fármacos que mimetizan la acción de la GLP1 endógena. Reducen efectivamente la HbA1c sin riesgo de hipoglicemia al aumentar la secreción de insulina en forma dependiente de glucosa y disminuir la secreción de glucagón. Además, producen baja de peso al retardar el VG y aumentar la saciedad. Actualmente hay cinco AR-GLP1 aprobados por la Food and Drug Administration (FDA) y seis por la European Medicines Agency (EMA) (Tabla 1). Todos se administran por vía subcutánea (sbc) y se diferencian en su origen, estructura molecular, farmacocinética, dosificación, administración, así como en su eficacia, tolerabilidad y satisfacción del paciente20. Se clasifican según sus características farmacocinéticas en agonistas de acción corta y de acción prolongada, pero en la actualidad, por el impacto que esto tiene en la práctica clínica, se separan también en prandiales y post prandiales según el mayor o menor efecto que ejercen sobre los niveles de glicemia en estado de ayuno o post ingesta21,22 (Tabla 1).
FÁRMACOS AGONISTAS DEL RECEPTOR DE GLP1 (AR-GLP1): CLASIFICACIÓN SEGÚN CARACTERÍSTICAS FARMACOCINÉTICAS Y ESTADO DE APROBACIÓN POR FDA Y EMA
| FÁRMACO | DURACIÓN ACCIÓN | EFECTO GLICÉMICO PREDOMINANTE | APROBACIÓN FDA* | APROBACIÓN EMA** |
|---|---|---|---|---|
| Exenatida | Corta | Prandial | 2005 | 2006 |
| Lixisenatida | Corta | Prandial | En trámite | 2013 |
| Liraglutida | Intermedia | Ayunas | 2010 | 2009 |
| Exenatida LAR | Prolongada | Ayunas | 2012 | 2011 |
| Albiglutida | Prolongada | Ayunas | 2014 | 2014 |
| Dulaglutida | Prolongada | Ayunas | 2014 | 2014 |
Exenatida (EXE) fue el primer AR-GLP1 aprobado para uso clínico en el año 2005. Es un producto sintético derivado de la exendina 4 (E4), un péptido de 39 aminoácidos (aa) que se obtiene a partir de la saliva del reptil Heloderma suspectum o monstruo de Gila, que tiene una estructura y un efecto biológico muy similar al GLP-1 humano. Es resistente a la acción de la DPP-4, lo que le confiere un efecto insulinotrófico 63% mayor que el GLP-1 nativo, una vida media plasmática de hasta 2,4 horas posterior a su administración sbc, y una duración total en plasma de hasta 10 horas. Se administra en dosis inicial de 5μg/2 veces al día, (60 minutos antes de dos comidas principales, con un lapso entre ambas no menor de 6 horas), y al mes de uso puede aumentarse a 10μg/2 veces según tolerancia23 (Tabla 2). En monoterapia reduce la HbA1c en 0,7 a 0,9%, e induce baja de 2,8-3,1kg de peso corporal24. La adición de EXE a pacientes no controlados con MF, SU o ambos, produce reducción dosis dependiente (0,5 o 10μg/2 veces/día) de HbA1c, glicemia en ayunas (GA) y peso, entre 0,4–0,86%, 5,4–10,8mg/dl y -0,9 a –2,8kg, respectivamente25, con mayor proporción de pacientes con HbA1c≤7% y similar incidencia de hipoglicemias leves comparada a placebo22,25. Así mismo ha demostrado mejoría glicémica y pérdida de peso asociada a TZD21. Por su parte en DM2 no controlados con SU+MF, la adición de EXE es similar en reducción de HbA1c comparada con insulina, pero con mejor control de la glicemia postprandial (GPP) y disminución del peso vs aumento de peso con insulina26,27. Seguimientos a 3-3,5 años han demostrado efectos favorables de EXE en biomarcadores hepáticos, factores de riesgo CV y mantención de la mejoría del control glicémico y disminución de peso respecto al estado basal28.
CARACTERÍSTICAS FARMACOLÓGICAS DE LOS AGONISTAS DE GLP1
| FÁRMACO | BASE PEPTÍDICA | VIDA MEDIA | T MÁX | DOSIS | ADMINISTRACIÓN |
|---|---|---|---|---|---|
| Exenatida | Exendina 4 | 2,4 hrs | 2,1 hrs | 5μg /2v día inicial 10μg /2v día máximo | 2 veces/día antes de las comidas |
| Lixisenatida | Exendina 4 | 2,7-4,3 hrs | 1,25-2,25 hrs | 10μg inicial 20μg máximo | 1 vez al día antes de la comida principal |
| Liraglutida | GLP1 | 11-15 hrs | 9 -12 hrs | 0,6mg inicial 1,8mg máximo | 1 vez al día sin relación con comidas |
| Exenatida LAR | Exendina 4 | 2,4 hrs | 2 semanas | 2mg semanal No requiere titulación | 1 vez/semana sin relación con comidas |
| Albiglutida | GLP1 | 6-8 días | 72-96 hrs | 30mg inicial 50mg máximo | 1 vez/semana sin relación con comidas |
| Dulaglutida | GLP1 | 5 días | 24-72 hrs | 0,75 mg 1,5mg máximo | 1 vez/semana sin relación con comidas |
Lixisenatida (LIXI) es un análogo sintético derivado de la E4. Se obtiene al eliminar la prolina en posición 38 y agregar 6 lisinas en el extremo terminal de E4, resultando una estructura de 44 aa con actividad biológica extendida y potencia 4 veces mayor que GLP1 humana29. A pesar de que su vida media es de 2,8hrs, tiene una acción prolongada que permite su administración una vez al día. Se cree que esto se debe a su gran afinidad por el receptor de GLP1 y a su marcada capacidad para retardar el VG29–31. La dosis inicial es de 10μg administrada una hora antes de la comida principal por dos semanas y luego se aumenta a 20μg/día (Tabla 2). Su eficacia y seguridad ha sido evaluada en los estudios GET GOAL (Glycemic Control and Safety Evaluation)30–32. En monoterapia disminuye la HbA1c en 0,85%, 47% de pacientes logran HbA1c<7%, con marcado efecto en la GPP, baja incidencia de hipoglicemias (1,7% vs 1,6% placebo), y efectos beneficiosos en peso corporal. Así mismo en pacientes no controlados con SU, MF o TZD, la adición de LIXI fue significativamente más efectiva que placebo en disminuir HbA1c, con mayor incidencia de hipoglicemias en asociación con SU. En un estudio comparativo de no inferioridad con EXE, (LIXI+MF vs EXE+MF), ambos análogos redujeron significativamente la HbA1c en 0,79% y 0,96% respectivamente, y aunque la diferencia entre ellos fue significativa, esta no excedió los márgenes de no inferioridad. Por otra parte en pacientes no controlados con insulina basal con o sin MF, la adición de LIXI disminuyó la HbA1c sin aumento de peso, redujo la dosis de insulina y la glicemia post prandial respecto a placebo31.
AR –GLP1 de acción largaExenatida LAR (Long-Acting Release) es una formulación de exenatida de liberación extendida que se obtiene por encapsulación de la droga activa en microesferas degradables, fabricadas con un copolímero de ácido láctico y ácido glicólico, que al inyectarse en el tejido sbc se hidrolizan y liberan el fármaco en forma lenta y progresiva, permitiendo su uso una vez por semana33. Se administran 2mg sbc cada 7 días, no requiere titulación progresiva de la dosis ni que ésta coincida con las comidas34 (Tabla 2).
Los estudios DURATION (Diabetes Therapy Utilization: Researching Changes in A1C, Weight and Other Factors through Intervention with Exenatide Once Weekly)15,34 demostraron superioridad de EXE semanal en comparación con EXE 2 veces/día en reducción de HbA1c y GA, y en proporción de pacientes con HbA1c<7%. Por su parte EXE de acción corta presentó mayor mejoría de la GPP. En monoterapia el control glicémico con EXE-LAR es muy similar al que se logra con MF o TZD, pero superior a sitagliptina (SITA). Así mismo agregado a MF es más efectivo en reducción de HbA1c y peso al compararse con SITA o TZD. En pacientes no controlados con MF+SU, la adición de EXE-LAR fue más efectiva que insulina en HbA1c, GPP y peso35.
Con EXE semanal la incidencia de hipoglicemias menores es baja (7%) y el mayor riesgo es con uso concomitante de una SU34. Resultados a largo plazo indican que las mejorías en los parámetros glicémicos y la pérdida de peso son mantenidas en el tiempo34.
Liraglutida (LIR) es un análogo sintético modificado de la GLP-1 humana que se obtiene por medio de tecnología ADN recombinante. Se sustituye un aa (lisina por arginina en la posición 34), y se incorpora una cadena de ácidos grasos en el espacio de la lisina en la posición 2636. Con estos cambios se logra una molécula con una estructura similar en un 97% a la GLP1 nativa, resistente a la DPP4, que tiende a auto-asociarse en el lugar de inyección y que en el plasma se une de manera no covalente a albúmina. Estas características permiten prolongar la vida media del análogo a 13 horas y la duración de su acción a 24 horas21,36. Liraglutida se administra vía sbc una vez al día en forma independiente de las comidas. La dosis inicial es de 0,6mg y se puede incrementar semanalmente según tolerancia hasta 1,8mg/día (Tabla 2).
Los estudios del programa LEAD (Liraglutide Effect and Action in Diabetes) que investigaron la efectividad y seguridad de LIR37–39, demostraron que en monoterapia reduce en forma dosis dependiente significativamente más que SU la HbA1c (-0,84% con 1,2mg y -1,14% con 1,8mg vs -0,51% con SU), con menor riesgo de hipoglicemia (8% vs. 24%) y con efectos favorables sobre el peso -2,1 y 2,5Kg. vs aumento de 1,1Kg con SU. En terapia combinada con SU reduce la HbA1c 1,1% vs placebo+0,2%, sin diferencias en el peso. Similar eficacia demostró la asociación a MF, pero con menor tasa de hipoglicemias. Al compararla con insulina glargina agregada a pacientes no controlados con SU+MF, LIR produjo una mayor reducción de HbA1c, con similar baja tasa de hipoglicemias. Finalmente, al comparar la eficacia de LIR 1,8mg/día o EXE 10mcg dos veces al día en pacientes con MF, SU o combinación de ambos, liraglutida resultó ser mas efectiva en control de HbA1c y con significativa mayor reducción de la GA. Por su parte EXE redujo más la GPP. La baja de peso fue similar con ambos fármacos (-3kg). Los resultados de los estudios del programa LEAD36,37 han mostrado que los efectos de liraglutida sobre la HbA1c, el bajo riesgo de hipoglicemia y la disminución del peso son mantenidos en el tiempo, y además con un perfil metabólico favorable sobre los lípidos y la presión arterial sistólica.
Albiglutida y Dulaglutida son nuevos agonistas de administración semanal aprobados en el año 2014 para el tratamiento de la DM221.
Albiglutida (ALBI) es un AR-GLP1 que contiene dos unidades de GLP1 modificadas por la sustitución de alanina por glicina, unidas a una molécula de albúmina humana. Su vida media es prolongada por resistencia a la DPP4 y a la unión a albúmina que enlentece la absorción desde el lugar de inyección, permitiendo administración semanal21,40 (Tabla 2). Los estudios HARMONY han demostrado que es superior a placebo, SITA y SU y similar a glargina y lispro en reducción de HbA1c. En comparación con LIRA es menos efectiva en control glicémico y reducción de peso40.
Dulaglutida (DUL) es un agonista que se obtuvo al unir dos moléculas modificadas de GLP-1 a un fragmento de inmunoglobulina humana resultando una proteína recombinante de alto peso molecular21,41. Estas características permiten una acción prolongada al enlentecer la absorción desde el sitio de inyección sbc y disminuir la tasa de eliminación renal. Su vida media es de 4,5 a 4,7 días según dosis de 0,75 o 1,5mg y se aplica con frecuencia semanal (Tabla 2). Los estudios AWARD (Assessment of weekly Administration of Dulaglutide) han demostrado superioridad de DUL en control glicémico vs placebo, MF, SITA, insulina y EXE de acción corta y no inferioridad en reducción de HbA1c comparada con LIRA. Disminuye HbA1c entre -0,78 a -1,51% y peso entre -0,35 a -3,03kg41.
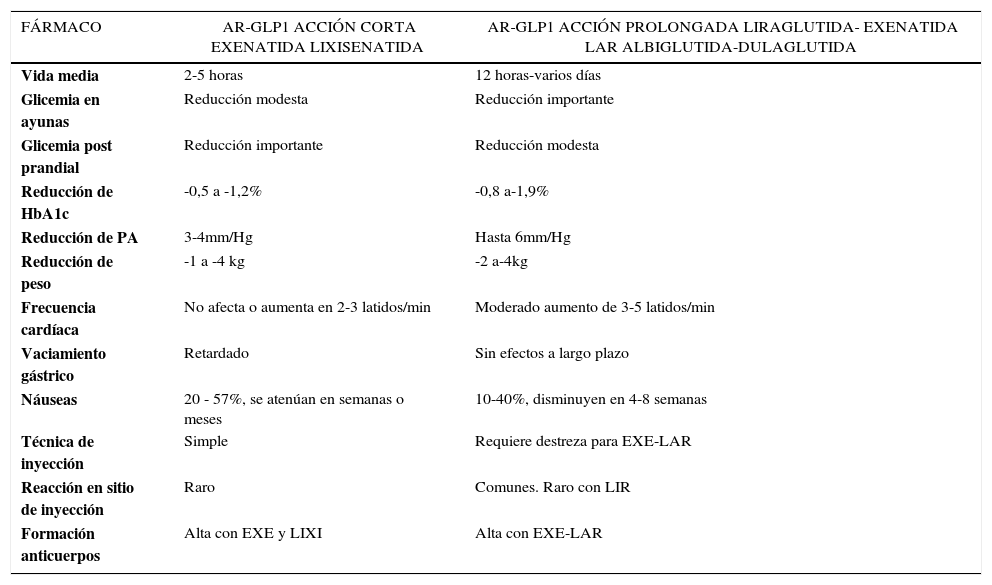
Eficacia comparativa de los AR-GLP1Control glicémico. Estudios de análisis comparativo de tipo aleatorio de los AR-GLP120,25,42 han demostrado que todos tienen alta eficacia en reducir la HbA1c ∼1%, sin embargo hay diferencias en la magnitud del efecto con un rango entre -0,60 a -1,9%. La mayor reducción de HbA1c se logra con LIR, seguido de EXE–LAR, EXE y LIXI (estos dos últimos con eficacia similar). También la efectividad de LIR es mayor que ALBI pero similar a DUL. Asimismo, el efecto sobre GA y GPP es diferente21,22. Mientras los AR-GLP1 de acción corta (EXE y LIXI) tienen ventajas en reducción de GPP, los de acción prolongada la tienen sobre la GA (Tabla 3). Esto se explica por el pronunciado retardo en el VG producido por los agonistas de acción corta. En contraste, la acción sobre el VG de los agonistas prolongados se pierde en el tiempo, lo que se atribuye a un fenómeno de taquifilaxis43 resultando en un menor impacto prandial30. El mayor efecto sobre la GA de los AR-GLP1 prolongados estaría mediado por una sostenida acción insulinotrópica e inhibitoria del glucagón por la activación continua de estos fármacos sobre el receptor de GLP142.
COMPARACIÓN DE ANÁLOGOS DE GLP1 DE ACCIÓN CORTA Y PROLONGADA
| FÁRMACO | AR-GLP1 ACCIÓN CORTA EXENATIDA LIXISENATIDA | AR-GLP1 ACCIÓN PROLONGADA LIRAGLUTIDA- EXENATIDA LAR ALBIGLUTIDA-DULAGLUTIDA |
|---|---|---|
| Vida media | 2-5 horas | 12 horas-varios días |
| Glicemia en ayunas | Reducción modesta | Reducción importante |
| Glicemia post prandial | Reducción importante | Reducción modesta |
| Reducción de HbA1c | -0,5 a -1,2% | -0,8 a-1,9% |
| Reducción de PA | 3-4mm/Hg | Hasta 6mm/Hg |
| Reducción de peso | -1 a -4 kg | -2 a-4kg |
| Frecuencia cardíaca | No afecta o aumenta en 2-3 latidos/min | Moderado aumento de 3-5 latidos/min |
| Vaciamiento gástrico | Retardado | Sin efectos a largo plazo |
| Náuseas | 20 - 57%, se atenúan en semanas o meses | 10-40%, disminuyen en 4-8 semanas |
| Técnica de inyección | Simple | Requiere destreza para EXE-LAR |
| Reacción en sitio de inyección | Raro | Comunes. Raro con LIR |
| Formación anticuerpos | Alta con EXE y LIXI | Alta con EXE-LAR |
Peso. Todos los AR-GLP1 producen baja de peso y en una magnitud significativamente mayor que la reportada con otros antidiabéticos42. Sin embargo, hay diferencias entre ellos. Las mayores bajas de peso se han observado con LIR y EXE de acción corta y prolongada. En los estudios confrontados, LIR produce mayor pérdida de peso frente a EXE (NS), y significativa frente a EXE-LAR, DUL, ALBI y LIXI20,42. La magnitud promedio de baja de peso es -2,9kg (-1 a -4Kg) (Tabla 3).
Seguridad y tolerabilidad de AR-GLP1Efectos adversos. Los efectos adversos más frecuentes, comunes a todos los AR-GLP1, se relacionan con distintos grados de intolerancia GI, especialmente náuseas, vómitos y diarrea. Las náuseas se presentan entre un 8-57% de los pacientes, suelen ser transitorias, dosis dependiente y disminuyen en tiempo variable (4-8 semanas) según el fármaco. Una estrategia para reducirlas es el aumento escalonado de la dosis30. Suelen presentarse con mayor intensidad con AR-GLP1 de acción corta lo que se relaciona con su mayor efecto enlentecedor del VG. Los estudios sugieren que son menos frecuentes con EXE-LAR en comparación con EXE y con LIR. A su vez, la frecuencia es similar entre LIR y EXE, pero son menos persistentes con el primero. Entre los agonistas de acción corta, LIXI produce menos estado nauseoso y por menor tiempo que EXE. ALBI tiene menor tasa de náuseas que LIR y esta última similar a DUL20,25,42Tabla 3.
Hipoglicemia: las tasas de hipoglicemia son bajas y similares entre los distintos grupos de agonistas. Son de carácter leve y se presentan fundamentalmente en pacientes tratados en forma concomitante con SU20.
Uso en insuficiencia renal (IR). No usar en pacientes con IR severa (FG<30ml/min). Pueden usarse con precaución (especialmente al inicio de la terapia y en etapa de escalamiento de dosis) con FG entre 30-50ml/min. Los efectos adversos GI inducidos por los agonistas pueden agravar la IR.
Reacciones en sitio de inyección. Los agonistas semanales se asocian a mayor incidencia de lesiones locales (nódulos ∼10%, prurito y eritema). La excepción parece ser dulaglutida. Entre los agonistas de uso diario, LIXI tiene más reacción local frente a EXE y aparentemente LIR tiene la menor incidencia de reacciones locales20,42.
Seguridad tiroidea. En roedores el uso de AR-GLP-1 se ha asociado a aumento de niveles de calcitonina y potencial desarrollo de tumores tiroideos. Sin embargo, no hay evidencia de una relación causal en humanos. En los estudios fase 3 con AR-GLP1 no se han demostrado mayores cambios en los niveles de calcitonina y hay solo un caso descrito de carcinoma papilar de tiroides que apareció en un paciente tratado con liraglutida42. Se recomienda no usar esta familia de fármacos en personas con antecedentes personales o familiares de cáncer tiroideo ni en portadores de neoplasia endocrina múltiple tipo 225,42.
Pancreatitis. Ha existido inquietud en la literatura respecto a los posibles efectos secundarios pancreáticos con el uso de AR-GLP1. En el año 2014 la FDA y la EMA en una declaración conjunta manifiestan que “los datos actualmente disponibles son inconsistentes con afirmar que existe una relación causal entre pancreatitis, cáncer de páncreas y el uso de fármacos con efecto incretina. Sin embargo, a pesar de que los datos analizados dan tranquilidad, hay incertidumbre respecto a la seguridad pancreática asociada a su uso a largo plazo. Por tanto se continuarán recopilando antecedentes y manteniendo las advertencias en las fichas técnicas de estos fármacos”44.
Inmunogenicidad. Se describe desarrollo de anticuerpos con EXE (27-49%), LIR (4,1-13%) y con LIXI (hasta un 70%). Sin embargo, no se ha encontrado impacto ni en la eficacia ni en la seguridad de estos fármacos25,42. La formación de Ac es muy baja en estudios fase 3 con DULA y ALBI42.
Seguridad cardiovascular (CV). Desde el año 2008 la FDA exige demostrar seguridad CV a los nuevos antidiabéticos, en estudios de no inferioridad con placebo efectuados en pacientes de alto riesgo CV45. Dado el efecto beneficioso demostrado por los AR- GLP1 sobre marcadores de disfunción endotelial y de riesgo CV, incluyendo disminución de PA, lípidos, peso, de la proteína C reactiva, del inhibidor del activador del plasminógeno tipo 1, del péptido natriurético cerebral y los posibles efectos directos sobre el miocardio y función ventricular se han creado expectativas respecto a un potencial rol protector de desarrollo de ECV en pacientes con DM218. En la actualidad hay varios ensayos que están evaluando la seguridad CV de los distintos agonistas. Recientemente se publicaron los resultados del estudio ELIXA46 realizado con LIXI, que demostró seguridad CV pero no superioridad en la reducción de eventos CV. En objetivos primarios (muerte CV, IAM, accidente cerebro vascular, hospitalización por angina inestable) no hubo diferencias en la tasa de eventos entre LIXI (13,4%) y placebo (13,2%). Asimismo, la hospitalización por insuficiencia cardíaca fue similar (4 y 4,2% respectivamente). Si bien estos resultados muestran neutralidad en riesgo CV, no se pueden extrapolar a los otros AR-GLP1 ya que los pacientes en estudio y los fármacos son diferentes. El estudio ELIXA se realizó en pacientes que habían sufrido síndrome coronario agudo; los otros ensayos en cambio se están efectuando en pacientes con cardiopatía coronaria estable u otros perfiles de alto riesgo CV. Por otra parte metaanálisis para valorar la seguridad CV que han comparado a los distintos agonistas con placebo u otros comparadores activos no han encontrado diferencias significativas en incidencia de eventos CV mayores entre los grupos47,48.
Efectos en el sistema nervioso central. Basados en los posibles efectos neuroprotectores de los AR-GLP1, estos fármacos se han propuesto como potenciales agentes terapéuticos en enfermedades neurodegenerativas. Hay varios estudios clínicos que están evaluando el posible rol terapéutico en enfermedad de Alzheimer, Parkinson y en trastornos cognitivos en pacientes con DM249.
Los AR-GLP1 están entre los fármacos más efectivos en reducir HbA1c, tienen efecto favorable sobre el peso y un mínimo riesgo de hipoglicemia. Su uso puede estar limitado por los efectos adversos (principalmente gastrointestinales), la administración subcutánea, y el costo20. Para mejorar la adherencia al tratamiento con estos fármacos, están en desarrollo varias alternativas de administración (vía oral, inhaladas y en dispositivos de liberación sbc continua que requeriría aplicación anual)50. Los AR-GLP1 están incorporados en los algoritmos terapéuticos de la DM2 como una opción a considerar en pacientes que no logran control glicémico y que requieren bajar de peso. Desde el año 2015 la Asociación Americana de Diabetes (ADA)51 los propone además como una alternativa a asociación con insulina basal en pacientes que requieren intensificación de la insulinoterapia. La adición de AR-GLP1 a insulina basal ha demostrado mejor control glicémico, menor riesgo de hipoglicemia y disminución de peso al compararse con insulina prandial52.
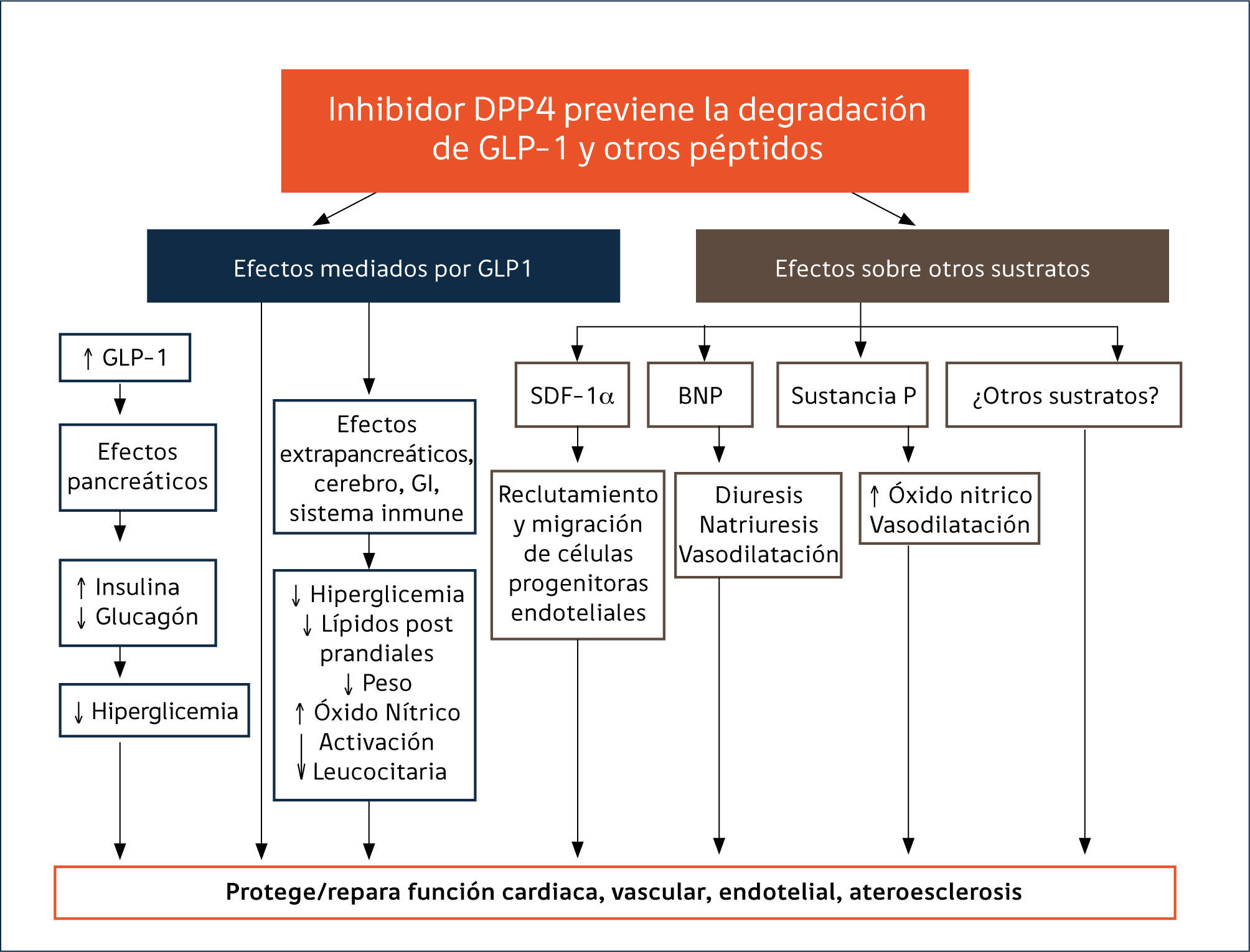
2. INHIBIDORES DE LA DIPEPTIDILPEPTIDASA 4 (IDPP-4)La dipeptidil peptidasa 4 (DPP-4) es una glicoproteína compleja que se encuentra ampliamente expresada en la superficie celular de varios tejidos y en forma soluble y activa en la circulación53,54. Esta aminopeptidasa, a través de degradación catalítica, inactiva a las incretinas intestinales GLP-1 y GIP, reduciendo su concentración y limitando sus acciones fisiológicas. Sin embargo, dada su amplia distribución, no es específica solo para las incretinas18,55. Se expresa además a nivel renal, páncreas, bazo, pulmón y en altos niveles en células endoteliales, epiteliales, dendríticas, inmunes T, linfocitos y macrófagos55. Participa, entre otros, en la regulación de la respuesta inflamatoria, en migración de células endoteliales, en las matrices colágenas, en funciones inmunológicas e interactúa con varios otros péptidos tales como factores de crecimiento, quimioquinas, neuropéptidos y péptidos vasoactivos18,54. Esta afinidad para una amplia gama de sustratos tiene el potencial para mediar una serie de efectos inmunológicos y pleiotrópicos CV, de manera dependiente e independiente de GLP-1, cuyo significado funcional no ha sido plenamente dilucidado18,55.
Existe cierta evidencia de que la actividad de la DPP-4 está aumentada en la DM256. Datos recientes muestran un aumento de la expresión de la enzima en tejido adiposo visceral y aumento de sus niveles circulantes en individuos obesos no diabéticos, planteando la posibilidad de que el aumento de la actividad de la DPP-4 puede jugar un rol fisiopatológico en las primeras etapas de desarrollo de la DM2 en el paciente obeso57. La identificación de la DPP4 como la enzima responsable de la vida media corta de las incretinas llevó a plantear que la inhibición de su acción, podría tener un rol en la terapia de la DM2, al aumentar los niveles de GLP1 y prolongar su acción insulinotrópica58. Estudios preclínicos y clínicos demostraron que la inhibición de la enzima mejoraba el control glicémico en pacientes diabéticos59, llevando al desarrollo de la familia de fármacos IDPP-4. Estos compuestos aumentan los niveles y potencian la acción de las incretinas endógenas, lo que lleva a aumento de la secreción de insulina y a disminución de la secreción de glucagón en forma dependiente de los niveles plasmáticos de glucosa53,58. En modelos animales se ha observado que, al igual que con los análogos de GLP-1, preservan la masa y la función de las células B de los islotes pancreáticos55. Sin embargo, dado que los aumentos de la GLP1 con los IDPP-4 son en niveles fisiológicos el impacto en el VG es menor60.
En la actualidad se encuentran aprobados numerosos inhibidores y otros están en diferentes fases de desarrollo61. Hay cinco IDPP-4 que están globalmente disponibles (sitagliptina (SITA) en uso clínico desde el 2006, vildagliptina (VILDA) 2007, saxagliptina (SAXA), 2010, linagliptina (LINA) 2011 y alogliptina (ALO) 2013, todos aprobados por la FDA excepto VILDA, aprobada por la agencia europea EMA61. Otros seis (anagliptin, evogliptin, gemigliptin, omarigliptin, teneligliptin, trelagliptin) están en uso solo en Corea y Japón61. Estos compuestos se diferencian en su estructura química y en sus propiedades farmacodinámicas y farmacocinéticas. Sin embargo, son similares en su eficacia hipoglicemiante y seguridad terapéutica58,61. Todos se administran por vía oral58,61. En esta revisión nos referiremos a los 5 primeros, de los cuales SITA, VILDA, SAXA y LINA se encuentran disponibles en Chile.
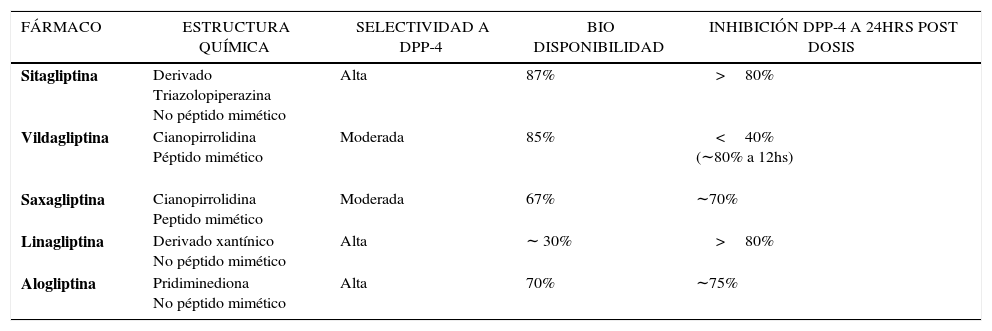
ESTRUCTURA QUÍMICA DE LOS IDPP-4Según su estructura química los potenciadores de incretinas se dividen en los que imitan a la molécula de la DPP-4 (peptidomiméticos) entre los que se encuentran VILDA y SAXA y los que no la imitan (no peptidomiméticos) SITA, LINA y ALO53,58,61 (Tabla 4). Todos producen inhibición competitiva y reversible en sustratos de la DPP-4 y tienen alta afinidad por la enzima, lo que resulta en una constante inhibición favoreciendo el control glicémico de ayuno y post prandial. Sin embargo, la forma de interacción con la enzima es diferente54,58,61. Los no peptidomiméticos tienen intrínsecamente una vida media larga, actúan en forma no covalente en el sitio catalítico del sustrato de la DPP-4 con acción inhibitoria sostenida por 24 horas. Por su parte, los peptidomiméticos tienen una vida media muy corta, que es prolongada por la fracción cianopirrolidina presente en estos fármacos58. Esta induce a una interacción covalente con la enzima dando lugar a uniones estables que determinan una acción inhibitoria más prolongada. Posteriormente, por medio de hidrólisis, se rompen los enlaces covalentes permitiendo una lenta liberación de la droga. De ahí que estos inhibidores sean conocidos también como de “lenta disociación”61. Debido a que la inactivación de la enzima por las distintas gliptinas es extra-celular, se conserva el funcionamiento de las principales proteínas intracelulares, lo que explica que no se afecten otras funciones atribuidas a las DPP-462.
ESTRUCTURA QUÍMICA Y CARACTERÍSTICAS FARMACODINÁMICAS DE INHIBIDORES DE DPP-4
| FÁRMACO | ESTRUCTURA QUÍMICA | SELECTIVIDAD A DPP-4 | BIO DISPONIBILIDAD | INHIBICIÓN DPP-4 A 24HRS POST DOSIS |
|---|---|---|---|---|
| Sitagliptina | Derivado Triazolopiperazina No péptido mimético | Alta | 87% | >80% |
| Vildagliptina | Cianopirrolidina Péptido mimético | Moderada | 85% | <40% (∼80% a 12hs) |
| Saxagliptina | Cianopirrolidina Peptido mimético | Moderada | 67% | ∼70% |
| Linagliptina | Derivado xantínico No péptido mimético | Alta | ∼ 30% | >80% |
| Alogliptina | Pridiminediona No péptido mimético | Alta | 70% | ∼75% |
Adaptado de: Deacon, C. F. and Lebovitz, H. E.
Diabetes, Obesity and Metabolism 2016; doi: 10.1111/dom.12610.
Selectividad. La DPP-4 es miembro de una familia de proteasas relacionadas genéticamente que incluye entre otras a la enzima DPP-8, DPP -9 y a la proteína activadora de fibroblastos α (FAPα)54,58,61. Hay evidencias que DPP-8 y -9 tienen un rol importante entre otros en la función inmune, hematopoyesis y protección de algunos parénquimas. Por su parte FAPα es una enzima extracelular que se expresa en fibroblastos y en regulación de tejidos de remodelación, generalmente ausente en el tejido adulto58. La relativa menor selectividad para DPP4, y mayor inhibición para DPP-8 y/o 9 podría asociarse a mayor posibilidad de efectos indeseables como disfunción inmune, trastornos hematológicos, alergias cutáneas, etc.54. En estudios preclínicos algunos prototipos de inhibidores se asociaron con distintos efectos tóxicos sugiriéndose que una probable menor inhibición a la DPP4 y más orientada a DPP-8 y/o 9 pudo haber sido responsable de los mismos63. Si bien, esto ha sido cuestionado, dio lugar a la optimización de los compuestos, mejorando la selectividad de la inhibición por la DPP-461 (Tabla 4). SITA y ALO son altamente selectivas. In vitro prácticamente no muestran actividad inhibidora frente a otros miembros de la familia DPP-4. LINA es también altamente selectiva con respecto a la DPP-8/9, pero menos respecto a la FAPα. Por su parte VILDA y SAXA son algo menos selectivas58,61. Sin embargo, aunque in vitro existen diferencias en la selectividad entre los diferentes IDPP-4 aprobados para uso clínico, no hay evidencia de efectos adversos relacionados con la inhibición de las otras enzimas cuando se utilizan en forma terapéutica61,64.
Absorción y distribución. Los IDPP-4 son moléculas pequeñas, no influenciadas por la ingesta, que posterior a su administración oral son rápidamente absorbidos, encontrándose una inhibición significativa de la enzima a los 5-15min de la dosis. La biodisponibilidad es alta (Tabla 4), se distribuyen ampliamente en el organismo y no atraviesan las membranas celulares58,65. La mayoría tiene muy baja unión a proteínas, a excepción de LINA que se une ampliamente a las proteínas plasmáticas54,58. No hay evidencia que los IDPP-4 crucen la barrera hematoencefálica, pero si atraviesan libremente la placenta58.
Metabolismo y eliminación. El 80% de la dosis de SITA, ALO y LINA se elimina inalterada. Las cantidades restantes se degradan a metabolitos inactivos o con muy baja afinidad a DPP-4. En contraste, VILDA y SAXA son extensamente metabolizados a nivel hepático. En el caso de VILDA a metabolitos inactivos y SAXA a compuestos que también son inhibidores competitivos reversibles de la DPP-4 con aproximadamente 50% de la potencia del fármaco original. Después de su administración circula un 25% de la droga original y 50% su metabolito activo61. Por su parte, la principal vía de eliminación de la mayoría de los IDPP-4 es el riñón58,61. En el caso de ALO y SITA, ésta es la única vía de eliminación. En contraste, la alta unión a proteínas de LINA determina que su depuración renal sea muy baja (<6%), y que se elimine casi en su totalidad por excreción biliar66 (Tabla 5). Estas diferencias en las vías de eliminación constituyen un criterio a considerar en la elección del tipo de IDPP-4 a usar en un determinado paciente61.
CARACTERÍSTICAS FARMACOCINÉTICAS DE LOS IDPP-4
| FÁRMACO | METABOLISMO | VÍA DE ELIMINACIÓN | VIDA MEDIA | DOSIS DIARIA RECOMENDADA |
|---|---|---|---|---|
| Sitagliptina | Mínimo | Predominantemente Renal (>80% droga activa) | ∼12½ h | 100mg 1 vez/día |
| Vildagliptina | Hepático independiente de CIP 450 Metabolito inactivo | Renal (22% fármaco original y 55% metabolito inactivo) | ∼2 h | 50mg 2 veces al día |
| Saxagliptina | Hepático vía CIP3A4/5 Metabolito activo | Renal (25% fármaco original y 55% metabolito activo) | ∼2½ h | 5mg 1 vez /día |
| Linagliptina | Mínimo | Predominantemente Biliar (< 6% renal) | ∼12 h | 5mg 1 vez /día |
| Alogliptina | Mínimo | Predominantemente Renal (>70% droga activa) | ∼21 h | 25mg 1 vez /día |
Adaptado de: Deacon, C. F. and Lebovitz, H. E.
Diabetes, Obesity and Metabolism 2016; doi: 10.1111/dom.12610
Vida media y eficacia inhibitoria. Los distintos IDPP-4 se diferencian también en la vida media y dosis terapéuticas58,61 (Tabla 5). Los no peptídicos por su interacción no covalente con la enzima tienen efecto casi inmediato, potente y de larga duración lo que permite su administración en una dosis diaria. En contraste los peptidomiméticos cuya acción requiere la formación de complejos inhibidores covalentes tienen acción corta, la que se prolonga por su lenta disociación. Esto se expresa en la práctica en que SAXA a pesar de tener una vida media de<5 horas se pueda utilizar una vez al día, y que VILDA pueda dosificarse dos veces al día61 (Tabla 5). Sin embargo, a pesar de las diferencias todos alcanzan a los 15 minutos de su administración un 90% de inhibición de la DPP-4, manteniéndola inactiva entre un 70-90% por 24 horas y aumentando entre 1,5 y 4 veces los niveles de GLP158,61.
Interacciones farmacológicas. Estos compuestos tienen mínimas interacciones farmacológicas, por lo que rara vez requieren ajustes de dosis con el uso concomitante con otros fármacos61. Sin embargo, dado su metabolismo hepático, la dosis de SAXA se debería disminuir si se usa con fármacos metabolizados por la isoenzima CYP3A4 del citocromo P450 (Ej. ketoconazol, claritromicina y antiretrovirales)18,61 (Tabla 6).
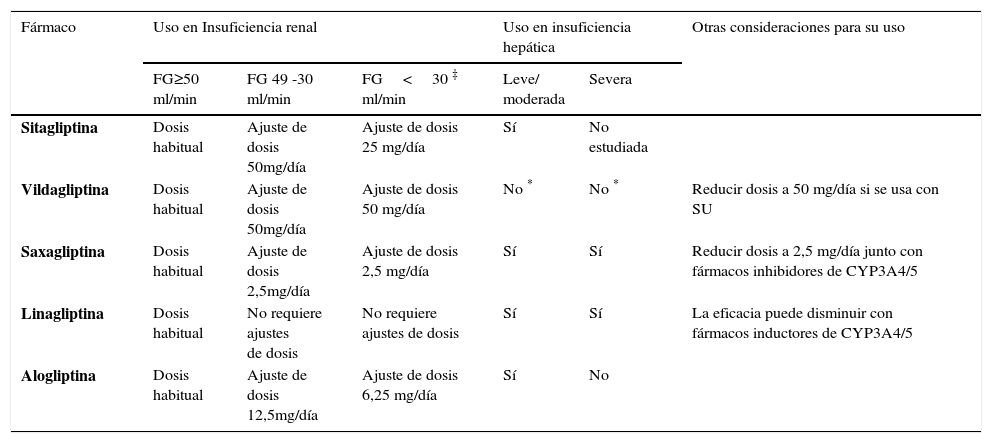
CONDICIONES CLÍNICAS QUE PUEDEN REQUERIR AJUSTES DE LA DOSIS DE ALGUNOS IDPP-4
| Fármaco | Uso en Insuficiencia renal | Uso en insuficiencia hepática | Otras consideraciones para su uso | |||
|---|---|---|---|---|---|---|
| FG≥50 ml/min | FG 49 -30 ml/min | FG<30 ‡ ml/min | Leve/ moderada | Severa | ||
| Sitagliptina | Dosis habitual | Ajuste de dosis 50mg/día | Ajuste de dosis 25 mg/día | Sí | No estudiada | |
| Vildagliptina | Dosis habitual | Ajuste de dosis 50mg/día | Ajuste de dosis 50 mg/día | No * | No * | Reducir dosis a 50 mg/día si se usa con SU |
| Saxagliptina | Dosis habitual | Ajuste de dosis 2,5mg/día | Ajuste de dosis 2,5 mg/día | Sí | Sí | Reducir dosis a 2,5 mg/día junto con fármacos inhibidores de CYP3A4/5 |
| Linagliptina | Dosis habitual | No requiere ajustes de dosis | No requiere ajustes de dosis | Sí | Sí | La eficacia puede disminuir con fármacos inductores de CYP3A4/5 |
| Alogliptina | Dosis habitual | Ajuste de dosis 12,5mg/día | Ajuste de dosis 6,25 mg/día | Sí | No | |
FG: Filtración glomerular
‡ Uso con precaución en pacientes con FG<15ml/min
Control glicémico. Revisiones sistemáticas y metaanálisis coinciden en demostrar que la eficacia de los distintos IDPP-4 es muy similar en monoterapia y en terapia combinada dual o triple, tanto en reducción de la HbA1c basal como en la proporción de pacientes que logran mejoría de HbA1c a cifras<7%67–69. En monoterapia reducen la GA en ∼18mg/dl (10-35mg/dl), la glicemia pp en ∼25mg/dL (20 a 60mg/dL) y la HbA1c en -0,7% (-0,4 a-1,0%)62. Sin embargo, hay muy pocos estudios de comparación directa entre los distintos IDPP-461. Un ensayo que comparó la eficacia de la adición de SAXA 5mg o SITA 100mg a pacientes no controlados con MF encontró que la reducción de HbA1c después de la adición de SAXA o SITA fue -0,52 y -0,62% respectivamente. Si bien el efecto de SAXA pareció ser más modesto, la diferencia entre los grupos no demostró inferioridad. Una proporción similar de pacientes logró HbA1c<6,5% (26 vs 29%), que era el objetivo a evaluar en el estudio70. Asimismo, en estudios comparativos a corto plazo, se ha demostrado que SITA y VILDA en monoterapia o asociadas a MF producen fluctuaciones diarias muy similares de los perfiles glicémicos71,72. También los estudios coinciden en que las mejores reducciones glicémicas con IDPP-4 se producen cuando la HbA1c inicial es mayor, lográndose mayor reducción con HbA1c inicial>9% vs<8%73.
Igualmente se ha comparado la eficacia de los IDPP4 con MF, TZD, AR-GLP1 y SU. Al respecto un metaanálisis comparativo en monoterapia de IDPP-4 vs MF encontró que los inhibidores se asociaron a significativa menor reducción de HbA1c y GA (diferencia promedio 0,28 (IC 95%0,17-0,40), y 0,81 (IC 95% 0,60-1,02)74. Comparadas con TZD no han demostrado inferioridad75. Por otra parte, los estudios que han comparado a los IDPP-4 con AR-GLP1, muestran la superioridad de los últimos en el control glicémico75,76. Sin embargo, hay controversia en la literatura en relación a la efectividad hipoglicemiante entre los IDPP-4 y las SU61. La ADA51 cataloga a los IDPP-4 como fármacos de potencia moderada (reducción HbA1c hasta 1%) y a las SU de alta potencia (reducción HbA1c 1,5%). Algunos afirman que estas diferencias no existen61 basados en estudios que han comparado en forma directa a los potenciadores de incretinas con SU [VILDA vs gliclazida77, SITA vs glibenclamida78 y LINA vs glimepirida79] que muestran uniformemente que la eficacia entre ambos tipos de fármacos es muy similar, y para ambas la reducción de HbA1c han sido menores a 1%. Se argumenta que la mayor efectividad que se le atribuye a las SU se debe a que se comparan “reducciones promedio” obtenidas con las diferentes clases de drogas en estudios que difieren en criterios de inclusión, tipos de población y niveles basales de HbA1c y no en resultados de estudios comparativos aleatorizados61. En segunda línea de terapia recientes metaanálisis coinciden en demostrar que la eficacia hipoglicemiante MF+IDPP-4 es inferior a la asociación MF+SU, con menor riesgo de hipoglicemia y menor ganancia de peso75,80. Es posible que cuando se conozcan los resultados del estudio GRADE (Glycemia Reduction Approaches in Diabetes: a comparative Effectiveness), que tiene como objetivo comparar efectividad, efectos adversos, durabilidad de la terapia etc., entre SU, IDPP4, AR-GLP1 y TZD asociados a MF81, se pueda definir si existen reales diferencias en la eficacia hipoglicemiante entre IDPP-4 y SU. En tercera línea de terapia un reciente metaanálisis que comparó todos los hipoglicemiantes orales e inyectables, incluyendo los IDPP-4 asociados a MF+SU, concluye que la eficacia es muy similar y las principales diferencias entre ellos se relacionan con el mayor o menor riesgo de hipoglicemias y efectos sobre el peso82.
Efectos sobre el peso: Estos fármacos tienen efecto neutro sobre en el peso corporal respecto a placebo y un efecto favorable cuando se les compara con SU (diferencia promedio -1,92Kg) o con TZD (-2,96Kg). Sin embargo, son inferiores en comparación con MF o AR-GLP-1 que se asocian a una reducción significativamente mayor de peso frente a IDPP-475. La adición de un IDPP-4 a cualquier otro hipoglicemiante no se asocia a cambios respecto al peso basal69.
SEGURIDAD TERAPÉUTICAHipoglicemia. Los IDPP-4, al aumentar la secreción de insulina de una manera dependiente de la glucosa, tienen muy bajo riesgo de producir hipoglicemia. Estudios comparativos en monoterapia o en terapia combinada demuestran en forma consistente que las tasas de hipoglicemia con las diferentes gliptinas es similar a placebo, MF y TZD y significativamente menor que con SU75,83. Sin embargo, la incidencia de hipoglicemias puede aumentar cuando se combinan con SU o insulina, de manera que en estos casos se recomienda reducir la dosis inicial de estos últimos61,83.
Efectos adversos. En general los inhibidores son fármacos muy bien tolerados58,61. Se han descrito algunas reacciones adversas GI, aunque su incidencia es muy baja. Con el uso inicial de SITA se describió un aumento de infecciones respiratorias altas y urinarias, lo que por algún tiempo fue motivo de preocupación dado el rol de las enzimas DPP-4 en la función inmune y el posible mayor riesgo de infecciones que pudieran ocurrir al inhibir su función58. Sin embargo, los metaanálisis recientes muestran que las tasas de infecciones de las vías respiratorias altas y urinarias son similares a placebo en estudios con VILDA, SAXA, LINA, y ALO. Con SITA y LINA existe un leve incremento de riesgo de nasofaringitis en comparación a placebo83. También se han reportado algunos casos de anafilaxis y angioedema asociados al uso de estos fármacos, aunque no está bien establecida una clara relación. En fecha reciente se comunicó que los inhibidores pueden provocar dolor articular grave que puede llegar a ser incapacitante84.
Pancreatitis. Desde el inicio del uso de potenciadores de incretinas se han comunicado casos de pancreatitis en pacientes tratados con estos fármacos, lo que ha llevado a plantear una posible asociación causal con patología pancreática. La evaluación de los datos disponibles no parece apoyar esta aseveración. Por otra parte en los recientes estudios de seguridad CV publicados con uso de IDPP-4 se han observado pocos casos de pancreatitis o cáncer pancreático y en una tasa similar o menor que placebo85–87. La FDA y la EMA consideran que el análisis de los datos actuales da tranquilidad y que se debe seguir evaluando la seguridad pancreática a más largo plazo44.
Seguridad cardiovascular (CV). La inhibición de la enzima DPP-4, junto con potenciar las acciones de la GLP1, modula la actividad de varios péptidos vasculares que se asocian a protección CV88 (Figura 1), creando expectativas por su potencial rol en la reducción de eventos CV en pacientes con DM2. El entusiasmo creció con los resultados de metaanálisis que incluían a SITA, VILDA, SAXA, LINA y ALO, en que estos inhibidores no se asociaron con un incrementado de eventos CV y algunos análisis sugerían una eventual protección88. Sin embargo, los estudios tenían limitaciones por su corta duración y su diseño no orientado a evaluar el impacto CV de estas drogas. Desde que la FDA recomendó estudios de seguridad CV para los nuevos fármacos45, se iniciaron los estudios SAVOR-TIMI (saxagliptina), EXAMINE (alogliptina), CARMELINA NCT01897532 (linagliptina), y TECOS (sitagliptina) comparando cada IDPP-4 vs placebo y el estudio CAROLINA NCT01243424 (linagliptina) con glimepirida como comparador activo. A la fecha, tres estudios han publicado sus resultados. El estudio SAVOR-TIMI85 que incluyó pacientes con ECV establecida o con múltiples factores de riesgo CV, el EXAMINE86 que estudió pacientes con síndrome coronario agudo reciente y el TECOS87 en pacientes con ECV establecida. Los tres estudios muestran de manera concluyente no inferioridad frente a placebo sobre eventos CV mayores (mortalidad por causa CV, IAM no fatal y ACV no fatal). Si bien no hay aumento de riesgo, tampoco existen beneficios CV asociados a su uso. SAXA mostró un incremento de riesgo significativo de hospitalización por insuficiencia cardíaca (IC) en comparación a placebo, (3,5% vs 2,8%;HR 1.27), aunque sin aumento de la mortalidad85. Posteriormente se demostró que el riesgo de IC fue más evidente durante los primeros 12 meses de terapia y mayor en los pacientes con condiciones de riesgo de IC, es decir, con antecedentes previos de falla cardíaca, niveles basales elevados de péptido natriurético (proBNP) o con enfermedad renal crónica89. Por otra parte, un análisis post hoc un del EXAMINE mostró una mayor tendencia NS de IC con ALO vs placebo (3,1 vs 2,9% HR 1,07, 95% CI 0,79-1,46), pero sin aumento de hospitalizaciones aún en pacientes con falla preexistente90. En el estudio TECOS87 la incidencia de IC con SITA fue idéntica a placebo (3,1%). Actualmente no hay explicaciones para el aumento de la incidencia de IC observada en el estudio SAVOR-TIMI61.
USO DE IDPP-4 EN SITUACIONES ESPECIALES
Insuficiencia renal (IR). Todos los IDPP-4 pueden utilizarse en pacientes con IR58,61. LINA, al no eliminarse por vía renal, es segura en cualquier etapa de falla y no necesita ajuste de dosis. No así las otras gliptinas, que requieren disminución de la dosis, especialmente SITA y ALO (Tabla 6). Cabe mencionar que las reducciones de dosis se basan fundamentalmente en la farmacocinética. Basados en su mecanismo de acción es poco probable que ocurran efectos adversos, ya que el inhibidor en sí no tiene efectos directos sobre la homeostasis de la glucosa. Más bien, los efectos serían secundarios a la inhibición de la DPP-4, pero, como en dosis terapéuticas la enzima es inhibida al máximo, cualquier aumento de la exposición a la droga no tiene mayor efecto61. De hecho, una serie de análisis de seguridad y grandes ensayos clínicos en pacientes con DM2 con daño renal en etapa terminal y diálisis, indican que los IDPP-4 son bien tolerados91.
Insuficiencia hepática (IH). La mayoría de estos fármacos pueden usarse en dosis terapéutica en pacientes con IH leve a moderada58. Aunque SAXA se metaboliza en el hígado, en casos de falla hepática aumenta solo ligeramente la exposición a la droga, lo que probablemente se debe a eliminación renal92. También, aunque la vía de eliminación de LINA es biliar, la IH no aumenta la exposición a la droga de forma clínicamente significativa. Se ha sugerido que la vía biliar de excreción es independiente de la función de metabolización hepática, y que bastaría con tener una función hepática mínima para mantener la eliminación de LINA y prevenir su acumulación93. Se ha informado una asociación entre uso de VILDA y aumento de enzimas hepáticas, pero esto no parece estar asociado a efectos adversos hepáticos. Sin embargo, VILDA está contraindicada en pacientes con cualquier grado de IH, incluyendo enzimas hepáticas elevadas pretratamiento61 (Tabla 6).
Adulto mayor (AM). Estudios clínicos efectuados con distintos IDPP-4 en AM>65 y>75 años coinciden en demostrar seguridad y eficacia tanto en monoterapia como en terapia asociada a otros agentes orales o insulina94–97. Por su parte en el importante subgrupo de AM del estudio SAVOR–TIMI se demostró seguridad CV, aunque también con un mayor riesgo de hospitalizaciones por IC99.
Los IDPP-4 han sido ampliamente aceptados en la práctica clínica, con un incremento progresivo en su uso100 debido a su seguridad, bajo riesgo de hipoglicemia, efectos neutros en el peso, fácil dosificación y administración oral. Su uso es especialmente ventajoso en las poblaciones más frágiles, como adultos mayores y pacientes con insuficiencia renal o mayor riesgo de hipoglicemia. Estos fármacos están incorporados en las diferentes guías clínicas en primera línea de terapia si hay contraindicación a MF, en asociación a MF o en triple terapia.
II. INHIBIDORES DEL COTRANSPORTADOR DE SODIO- GLUCOSA TIPO 2 (ISLGT2)La participación del riñón en la homeostasis de la glucosa fue descrita por primera vez en 1930101. Sin embargo, a pesar de la gran cantidad de evidencia acumulada en los años siguientes, no se le dio mayor importancia hasta que se conoció que alteraciones en mecanismos renales de regulación de la glucosa estaban involucradas en la patogenia de la DM2. Así, este órgano se transformó en centro de investigación como otro objetivo terapéutico para el manejo de la hiperglicemia, dando origen a los fármacos inhibidores del transportador Na+−glucosa Tipo 2 (ISLGT2), que reducen los niveles glicémicos al aumentar la excreción renal de glucosa en forma independiente de la insulina101,102.
Rol del riñón en la homeostasis de la glucosa y en la hiperglicemia de la DM2El riñón participa en la regulación de la glucosa a través de gluconeogénesis (contribuyendo al 20−25% de los niveles de glicemia en estado de ayuno), filtración, reabsorción y consumo de glucosa101. En condiciones fisiológicas el glomérulo filtra ∼180g de glucosa al día, cantidad que posteriormente se reabsorbe casi en su totalidad a nivel tubular, eliminándose por la orina<1% de la glucosa filtrada. La reabsorción es mediada por transportadores de glucosa SGLT (Sodium−Glucose Linked Transporter) que forman parte de una familia de seis proteínas de membrana que están ampliamente distribuidos en el organismo de los cuales dos, SGLT1 y SGLT2, se expresan en el túbulo contorneado proximal (TCP). Ambos se diferencian en su ubicación y en la afinidad y capacidad de transporte de glucosa101,103. SGLT1 es un transportador de baja capacidad y alta afinidad por la glucosa que se expresa fundamentalmente a nivel intestinal, donde es el principal transportador de glucosa/galactosa. También se encuentra, aunque en menor cantidad, en los segmentos S2 y S3 del TCP donde reabsorbe ∼10% de la glucosa. Por su parte SGLT2 es un trasportador de alta capacidad y baja afinidad, con expresión casi exclusiva en la superficie luminal del segmento S1 del TCP, donde reabsorbe el 90% de la glucosa filtrada por el riñón. La reabsorción de glucosa por los transportadores SLGT se produce por medio de un proceso de cotransporte activo Na+−glucosa, en que el Na+ al absorberse a través de la membrana celular crea una gradiente de energía que permite el ingreso de la glucosa en forma independiente de la insulina. Desde la pared tubular, ambos reingresan a la circulación, la glucosa vía transportadores de glucosa GLUT1 y GLUT2 y el Na+ por medio de la bomba Na+−KATPasa.102,104. La glucosa filtra libremente por el glomérulo, de modo que, si los niveles de glicemia aumentan, la cantidad de glucosa filtrada aumenta en forma lineal. Sin embargo, la capacidad de reabsorción del sistema de transporte SGLT renal es limitada con un umbral de saturación a valores de glicemia de ∼180−200mg/dl según variaciones individuales. Toda la glucosa filtrada por encima de este umbral supera la velocidad de transporte máximo que es de ∼375ml/min, y es eliminada por la orina102,104. Si se considera que el riñón diariamente reabsorbe 180g/glucosa, produce 15 a 55g y metaboliza 25−35g, la reabsorción tubular constituye el mecanismo más importante a través del cual los riñones influencian la homeostasis de la glucosa102,105.
En la DM2 existen alteraciones en el manejo renal de la glucosa que contribuyen a la hiperglicemia9,102. Hay defectos en la gluconeogénesis, lo que resulta en una mayor producción de glucosa, y aumento de la expresión y la capacidad de absorción de los SLGT2. Esta sobreexpresión de SLGT2 aumenta el transporte máximo (Tm) para la glucosa a valores 40-60mg/dl más altos que el umbral fisiológico de saturación (glicemia±180mg/dl en sujetos sanos a 240mg/dl en DM2) de manera que la glucosuria se inicia a partir de niveles glicémicos más elevados a los esperados, lo que se traduce en una mayor reabsorción de glucosa contribuyendo a mantener la hiperglicemia101,102,106. Existen mutaciones genéticas que producen trastornos funcionales de los transportadores. Defectos en SLGT1 originan un síndrome de mala absorción con severa diarrea y mínima glucosuria, lo que muestra el predominio de expresión intestinal de este transportador. Por su parte, mutaciones de SLGT2 causan glucosuria renal familiar y a pesar que ésta puede llegar a ser muy elevada, no se asocia a desarrollo de daño renal, vesical, infecciones de las vías urinarias ni hipoglicemias. El conocimiento de la sobreexpresión de SLGT2 en la DM2 sumado a que la inhibición “natural” del mismo es de curso benigno llevó al desarrollo de fármacos inhibidores del transportador tubular de glucosa SLGT2104,106,107.
Fármacos inhibidores de la SLGT2 (ISLGT2)A mediados del siglo XIX se describió que la florizina, una sustancia extraída de la corteza del manzano, originaba glucosuria108. El interés en el compuesto se inició en los años 50, al demostrarse que bloqueaba el transporte de glucosa en varios tejidos, incluido el riñón, lo que motivó que posteriormente se usara para estudiar la función de los transportadores SLGT. Se comprueba que bloquea los SGLT1 y SGLT2, que produce glucosuria, disminuye la hiperglicemia, pero además genera un síndrome de malabsorción, por su mayor afinidad a SLGT1. Cuando se demostró que la excesiva reabsorción de glucosa por el riñón tenía un rol patogénico en la DM29,102, se evaluó la florizina con fines terapéuticos; sin embargo, su uso no fue posible por efectos adversos y baja biodisponibilidad. La investigación se orientó a derivados de la florizina109, obteniéndose compuestos con potente selectividad a SGLT2 con mayor estabilidad, mejor biodisponibilidad y con adecuada tolerancia dando origen a este nuevo grupo de fármacos inhibidores selectivos de SGLT2 (ISLGT2), con principios terapéuticos totalmente diferente al de otros hipoglicemiantes110. Estos compuestos inhiben en forma competitiva, reversible y selectiva al cotransportador SLGT2 ubicado en el TCP; su acción es independiente de la secreción o acción de la insulina y de la etapa de evolución de la DM2102,106. La inhibición “resetea” el sistema reduciendo el umbral de saturación para la glucosuria de manera que ésta se inicie con niveles de glicemia 60-80mg menores (∼100mg/dl), con lo que aumenta la excreción urinaria de glucosa en 60-80g/día102. Como resultado de la glucosuria farmacológica se reduce la hiperglicemia y adicionalmente se produce baja de peso debido a la pérdida de calorías (4kcal/g de glucosa excretada) asociada a la mayor cantidad de glucosa eliminada, pudiendo llegar a perderse 240-320cal/día102,106. Actualmente hay tres fármacos ISLGT2 aprobados por la FDA y la EMA: canaglifozina (CANA) (2013), dapagliflozina (DAPA) (2014) y empagliflozina (EMPA) (2014). Otros, ipragliflozin, tofogliflozina y luseogliflozin están en uso en Japón, y aún en desarrollo (ertugliflozin, remogliflozin y sotagliflozina)105,111.
CANA, DAPA y EMPA son derivados C glucósidos de la florizina. De los tres, EMPA es el más selectivo para SLGT2 (Tabla 7). Todos se administran por vía oral, tienen alta biodisponibilidad y una vida media ≥12 horas, lo que permite dosificarlos una vez al día. Aunque pueden administrarse con o sin alimentos, se recomienda que CANA se use antes de la primera comida del día. No tienen interacciones farmacológicas clínicamente relevantes y pueden usarse en monoterapia o en asociación con cualquier otro hipoglicemiante, incluyendo insulina. Su uso requiere una adecuada función renal (Tabla 7) y pueden usarse sin ajuste de dosis en casos de insuficiencia hepática leve a moderada. No hay estudios de seguridad en insuficiencia hepática severa111. La efectividad de estos compuestos en reducción de HbA1c es dosis dependiente (Tabla 8), su uso no se asocia a hipoglicemia y, en forma adicional, producen baja de peso, disminución de la PA y un modesto aumento de ∼5% de colesterol HDL, condiciones consideradas un factor agregado favorable para pacientes con sobrepeso e HTA. Por otra parte, con este grupo farmacológico se ha descrito mejoría tanto de la función de la célula β como de la sensibilidad a la insulina. Si bien estos fármacos no afectan directamente la secreción de insulina ni la captación de glucosa en los tejidos, tienen un impacto beneficioso en ambas condiciones al corregir la hiperglicemia y por ende, la glucotoxicidad102,112.
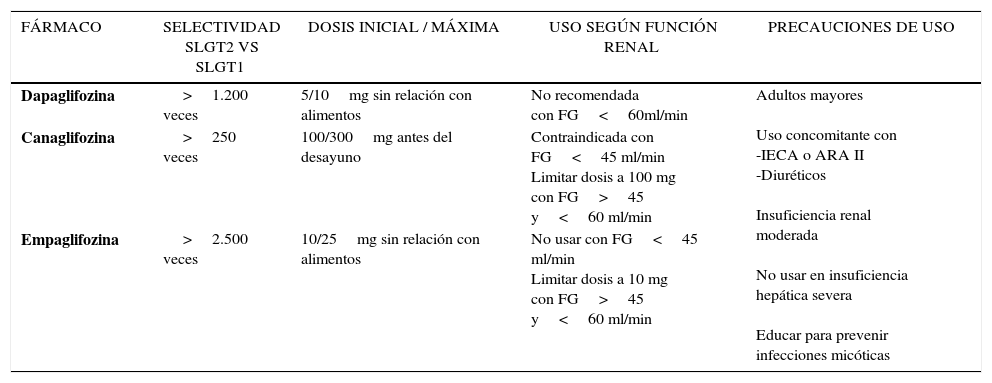
SELECTIVIDAD Y CONSIDERACIONES PARA EL USO CLÍNICO DE LOS INHIBIDORES DEL TRANSPORTADOR SODIO-GLUCOSA TIPO 2 (ISLGT2)
| FÁRMACO | SELECTIVIDAD SLGT2 VS SLGT1 | DOSIS INICIAL / MÁXIMA | USO SEGÚN FUNCIÓN RENAL | PRECAUCIONES DE USO |
|---|---|---|---|---|
| Dapaglifozina | >1.200 veces | 5/10mg sin relación con alimentos | No recomendada con FG<60ml/min | Adultos mayores Uso concomitante con -IECA o ARA II -Diuréticos Insuficiencia renal moderada No usar en insuficiencia hepática severa Educar para prevenir infecciones micóticas |
| Canaglifozina | >250 veces | 100/300mg antes del desayuno | Contraindicada con FG<45 ml/min Limitar dosis a 100 mg con FG>45 y<60 ml/min | |
| Empaglifozina | >2.500 veces | 10/25mg sin relación con alimentos | No usar con FG<45 ml/min Limitar dosis a 10 mg con FG>45 y<60 ml/min |
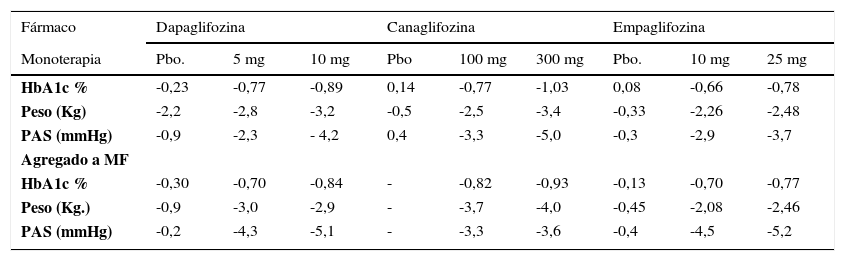
EFICACIA EN CONTROL GLICÉMICO, PESO Y PRESIÓN ARTERIAL SISTÓLICA (PAS) CON DAPAGLIFOZINA, CANAGLIFOZINA Y EMPAGLIFOZINA. RESUMEN DE ESTUDIOS CLÍNICOS DE MÁS DE 24 SEMANAS DE OBSERVACIÓN
| Fármaco | Dapaglifozina | Canaglifozina | Empaglifozina | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Monoterapia | Pbo. | 5 mg | 10 mg | Pbo | 100 mg | 300 mg | Pbo. | 10 mg | 25 mg |
| HbA1c % | -0,23 | -0,77 | -0,89 | 0,14 | -0,77 | -1,03 | 0,08 | -0,66 | -0,78 |
| Peso (Kg) | -2,2 | -2,8 | -3,2 | -0,5 | -2,5 | -3,4 | -0,33 | -2,26 | -2,48 |
| PAS (mmHg) | -0,9 | -2,3 | - 4,2 | 0,4 | -3,3 | -5,0 | -0,3 | -2,9 | -3,7 |
| Agregado a MF | |||||||||
| HbA1c % | -0,30 | -0,70 | -0,84 | - | -0,82 | -0,93 | -0,13 | -0,70 | -0,77 |
| Peso (Kg.) | -0,9 | -3,0 | -2,9 | - | -3,7 | -4,0 | -0,45 | -2,08 | -2,46 |
| PAS (mmHg) | -0,2 | -4,3 | -5,1 | - | -3,3 | -3,6 | -0,4 | -4,5 | -5,2 |
*Pbo: Placebo
Adaptado de ref. 106 y 111.
Fujita Y and Inagaki N. Renal sodium glucose cotransporter 2 inhibitors as a novel therapeutic approach to treatment of type 2 diabetes: Clinical data and mechanism of action. J Diabetes Invest 2014; 5: 265–275 y Handelsman Y. Potential Place of SGLT2 Inhibitors in Treatment Paradigms for Type 2 Diabetes Mellitus Endocr Pract. 2015; 21:1054-1065
Control glicémico. Los ISLGT2 son efectivos en reducir la glicemia de ayuno, postprandial y la HbA1c. Diferentes estudios han evaluado su eficacia en monoterapia vs placebo, en combinación con MF, o agregados a otros hipoglicemiantes. Estudios de 24 semanas o más en monoterapia106,111 muestran que los distintos ISLGT2 son eficaces en reducción de HbA1c, con una aparente superioridad de CANA (Tabla 8). Sin embargo, un metaanálisis que evaluó a los diferentes ISLGT2 encontró que los estudios con CANA tenían una mayor heterogeneidad lo que no permite afirmar que su eficacia hipoglicemiante sea superior113. Por otra parte otro metaanálisis que incluye 45 estudios en que se compara a los ISLGT2 vs placebo en monoterapia y 13 estudios con ISLGT2 vs comparadores activos (MF, SITA, y SU), muestra que en monoterapia reducen la HbA1c-0,79% (-0,96 a -0,62) vs placebo, asociados a otro hipoglicemiante -0,61% (-0,69 a -0,53) vs placebo, y al compararse con otro hipoglicemiante en monoterapia o en asociación vs placebo son similares con diferencias de HbA1c de 0,05% (-0,06 a 0,16) y 0,16% (0,32 a 0,00) respectivamente114.
Efectos en el peso. La pérdida de calorías secundaria a la glucosuria determina que todos los ISLGT2 produzcan baja de peso en forma independiente a su uso en monoterapia o en asociación114,115 (Tabla 8). La reducción inicial es mayor y se hace evidente después de aproximadamente 6 semanas de uso con -2,2 a -3,4Kg, siendo probable que en esto también influya la pérdida de líquidos por la diuresis osmótica que producen estos fármacos115. A los 6-12 meses de terapia la pérdida de peso se estabiliza en ∼-2kg, cifra considerada menor a la esperada, lo que sugiere que esta forma de tratamiento estimularía la ingesta calórica112,114.
Efectos en la presión arterial (PA). Con estos fármacos se produce una importante reducción de la PA sistólica (PAS) y diastólica (PAD)51 (Tabla 8). Un metaanálisis de 27 estudios aleatorizados que evaluaron el impacto de los ISLGT2 en la PA encontró una diferencia promedio de la PAS de -4,0mmHg y de la PAD -1,6mmHg, al compararlas con el valor inicial116. Resultados similares se han reportado en otros estudios114. Es probable que la disminución de la PA se deba a la diuresis osmótica, pero también la leve pérdida de peso puede contribuir. Se ha hecho notar que esta disminución de la PA no se asocia a aumento en la frecuencia cardíaca115.
SEGURIDAD EN USO CLÍNICOEn términos de seguridad, los estudios con DAPA, CANA y EMPA, han demostrado que son fármacos bien tolerados y con un perfil de seguridad favorable. Sus efectos adversos se relacionan principalmente con la glucosuria propia de su mecanismo de acción y son de carácter leve y muy rara vez obligan a abandonar el tratamiento111,114.
Hipoglicemia. El uso de estos compuestos se asocia con un bajo riesgo de hipoglicemia lo que se atribuye por una parte a su mecanismo de acción independiente de la insulina y a que la glucosuria se inicia por encima de los niveles glicémicos que producen síntomas hipoglicémicos117. El riesgo de hipoglicemia es similar o ligeramente mayor a placebo, similar a la MF y aumenta con uso concomitante de SU o insulina114,115.
Depleción de volumen. Los ISLGT2 causan una modesta diuresis osmótica y poliuria lo que puede provocar síntomas relacionados con depleción de volumen como hipotensión, síncope y falla renal. Estas condiciones pueden afectar especialmente a adultos mayores que no regulan adecuadamente la ingesta hídrica118, a pacientes con insuficiencia renal o quienes están en terapia concomitante con diuréticos de asa o con inhibidores del sistema renina-angiotensina aldosterona, situaciones en que hay que ser especialmente cuidadoso con el uso de estos fármacos51,111,119.
Infecciones genitourinarias. El efecto adverso más frecuente son las infecciones micóticas genitales y las infecciones urinarias (ITU)111. Un metaanálisis, que ha incluido la mayor cantidad de estudios clínicos, encontró con ISLGT2 un aumento significativo de infecciones micóticas (OR 5,06, IC95%=3,44-7,45) y de ITU(OR1,42, IC95%=1,06-1,90) vs comparadores activos114. Ambas infecciones suelen ser de carácter leve a moderado y revierten con terapia habitual120. Se presentan especialmente en etapas tempranas del tratamiento y no son recurrentes. El mayor riesgo de ITU es en mujeres y las infecciones micóticas se presentan más en mujeres con antecedentes de micosis genital y en hombres no circuncidados51,111.
Seguridad cardiovascular (CV). Los ISLGT2, más allá del control glicémico, modifican positivamente una serie de factores de riesgo CV como la PA, el peso, la hiperinsulinemia, albuminuria, adiposidad visceral y stress oxidativo, lo que representa un importante potencial para reducir la ECV del paciente con DM111,121. Por otro lado, su uso se asocia a pequeños aumentos de las lipoproteínas de alta densidad (HDL) y de baja densidad (LDL), con disminución de los triglicéridos, cuya importancia clínica no está establecida y que teóricamente podría opacar algunos de estos beneficios121. El impacto CV de estos fármacos está en evaluación con CANA (estudio CANVAS NCT0103262) y con DAPA (estudio DECLARE NCT01032629). Recientemente se publicaron los resultados del estudio de seguridad cardiovascular EMPA-REG122 que comparó EMPA vs placebo en pacientes DM2 de alto riesgo CV. No se encontraron diferencias entre los grupos en las tasas de IAM o ACV no fatal; sin embargo, con EMPA hubo una reducción significativa del 38% del riesgo relativo de muerte por causa CV, una reducción significativa del 32% en todas las causas de mortalidad, y una reducción significativa del 35% de hospitalización por IC, convirtiéndose en el primer estudio publicado con nuevos fármacos que demostró un impacto CV favorable en pacientes de alto riesgo vascular. Aún no hay una clara explicación para los resultados. Por un lado se piensa que el efecto de EMPA no es sobre la enfermedad ateroesclerótica, ya que de ser así habría disminuido el IAM y el ACV. Se cree que podría deberse a efectos asociados a la diuresis osmótica, lo que explicaría la disminución de IC y sus complicaciones asociadas que parecen dar cuenta de los mayores beneficios del estudio123. Se discute si es un efecto de clase, lo que probablemente se definirá con los resultados de los próximos estudios. Por otra parte estos resultados favorables son en un grupo de pacientes muy específicos, con enfermedad CV establecida, de manera que no son necesariamente extrapolables a todos los pacientes con DM2.
Uso en insuficiencia renal. Dado que el sitio de acción de los ISLGT2 es el riñón, estos fármacos requieren de una adecuada función renal para ser efectivos. No deben usarse con FG<45ml/min o diálisis51,115,123. Cada compuesto tiene un punto de corte para iniciar o ajustar dosis según función renal (Tabla 7). Se ha descrito una ligera y reversible disminución de la FG en las primeras semanas de terapia con ISLGT2 que, en la mayoría de los casos, no requiere suspensión del fármaco; sin embargo, se debe tener especial cuidado si existen condiciones que favorecen la depleción de volumen. El uso de estos fármacos se ha asociado a disminución de albuminuria, postulándose un posible rol protector renal que va más allá de lo esperado por la reducción de la glicemia y PA. Se explicaría por cambios asociados a la inhibición de la reabsorción tubular de Na+que resultan en disminución de la presión intraglomerular y por tanto de la hiperfiltración que es uno de los principales mecanismos responsables de la nefropatía DM102,104,121. Esta posibilidad está actualmente bajo investigación (CANVAS R, NCT01989754 y CREDENCE, NCT02065791).
Seguridad ósea. Está en evaluación si el uso de estos fármacos se asocia a alteraciones en el metabolismo óseo. Por su mecanismo de acción podrían alterar la absorción de calcio y fosfatos y tener consecuencias en el hueso. Hay antecedentes de aumento de fracturas óseas con DAPA en pacientes con insuficiencia renal114,124 y en un análisis de seguridad con CANA se habría encontrado un aparente aumento de la incidencia de fracturas frente a placebo125. Sin embargo, no se han encontrado alteraciones en marcadores de recambio óseo ni de densidad ósea con estos fármacos126. Se ha recomendado estar alertas125 y evaluar esta condición en los estudios clínicos. En el estudio EMPA-REG la tasa de fracturas fue baja e idéntica a placebo122.
Cetoacidosis (CAD). Se han informado algunos casos serios de CAD en pacientes en tratamiento con ISLGT2. Algunos reportes han sido en DM1 o LADA, condiciones en que el uso de estas drogas no está aprobado. Pero también se ha comunicado en DM2, y si bien en algunos casos se ha presentado en forma concomitante con una patología aguda, o reducción de la dosis de insulina, en otros no se ha logrado identificar un factor causal. La presentación de la cetoacidosis en DM2 puede ser atípica, con euglicemia, lo que dificulta su diagnóstico. La FDA127 ha alertado sobre este riesgo; sin embargo, un comité de expertos de la Asociación Americana de Endocrinólogos Clínicos y del Colegio Americano de Endocrinólogos (AACE/ACE), concluyó que la incidencia de CAD es poco frecuente y el riesgo-beneficio favorece el uso de los ISGLT2 sin cambios en sus recomendaciones actuales123,128.
En suma los ISLGT2 son fármacos con un novedoso mecanismo de acción, bien tolerados y eficaces en el tratamiento de la DM2. No producen hipoglicemia y ofrecen beneficios adicionales tales como pérdida de peso, reducción de la presión arterial y aumento de HDL. Son el primer grupo de los nuevos fármacos en que uno de sus compuestos muestra beneficios CV en pacientes de alto riesgo vascular. A pesar que son los fármacos más recientemente aprobados para uso clínico han sido incorporados en las guías clínicas51,123 como monoterapia y especialmente como una interesante opción a considerar en segunda o tercera línea del algoritmo terapéutico de la DM2.
CONCLUSIÓNLa DM2 es una enfermedad progresiva que habitualmente requiere de 2 o más fármacos para lograr un adecuado control. En las recomendaciones de tratamiento farmacológico de la DM2 hay consenso en que la terapia inicial es la MF a menos que esté contraindicada y cuando con esta no se logra control, está indicada la terapia dual. La ADA/EASD51 recomienda que la elección del segundo o tercer fármaco sea centrada en el paciente y se consideren las comorbilidades, el riesgo de hipoglicemia, el peso, los potenciales efectos secundarios, la eficacia, el costo, y las preferencias del paciente, sin recomendar ningún fármaco de los actualmente disponibles sobre otro. No así la AACE/ACE123 que, en su reciente consenso, privilegia la seguridad terapéutica estableciendo como primeras opciones a asociar a MF a los nuevos fármacos AR-GLP1, ISLGT2 o IDPP-4, por su bajo riesgo de hipoglicemia, sus efectos positivos o neutros sobre el peso y, en el caso de los ISLGT2, por sus adicionales efectos beneficiosos sobre la PA.