






En la edad pediátrica las epilepsias son frecuentes, con manifestaciones clínicas diversas y edad-dependientes. El reconocimiento preciso de los diferentes síndromes y epilepsias es importante para determinar un tratamiento y pronóstico adecuados. Cerca de un 70% de las epilepsias en niños tiene una buena respuesta a tratamiento médico o remisión espontánea y buen pronóstico en el largo plazo; se agrupan aquí las formas idiopáticas ya sea generalizadas como focales. Un grupo menor, pero no menos importante, es refractario a fármacos antiepilépticos, destacando las Encefalopatías Epilépticas que implican un mal pronóstico neurológico y un grupo de pacientes refractarios que serán candidatos a otras alternativas de tratamiento, como cirugía de la Epilepsia. Se revisan los principales conceptos que subyacen a la actual clasificación de Síndromes Electroclínicos y Epilepsias de ILAE y las características de algunos síndromes electro-clínicos atendiendo a su frecuencia, relevancia de un diagnóstico preciso y aportes recientes de significación
Epilepsy is a common condition in the pediatric age, with age-dependent and heterogeneous clinical manifestations. A successful approach to treatment and prognosis requires the precise recognition of the diverse syndromes and epilepsies. Near 70% of pediatric epilepsies, especially de idiopathic focal and generalized syndromes, are responsive to medical treatment or have spontaneous remission and a positive longterm prognosis. A smaller but not less important group is refractory to antiepileptic drugs and will need other treatment options such as epilepsy surgery. Here we discuss the main concepts underlying the ILAE Classification of Electroclinical syndromes and Epilepsies and the characteristics of some syndromes attending to its frequency, relevance of precise diagnosis and recent advances.
La Epilepsia o las Epilepsias son un complejo sintomático, con diversidad de etiología, características clínicas, electrofisiológicas, de imágenes y patológicas. En la edad pediátrica se distingue diferentes Epilepsias y Síndromes Epilépticos cuyo denominador común es la predisposición a presentar crisis recurrentes, sin embargo con muy diferente pronóstico, respuesta a tratamiento y comorbilidades. Su diagnóstico acucioso es clave para desarrollar una adecuada estrategia de manejo y tratamiento y así proporcionar la mejor calidad de vida a nuestros pacientes. Para ello es necesario identificar el(los) tipo(s) de crisis, información que se conjugará con otras características clínicas como edad de inicio, condición neurológica, factores etiológicos, antecedentes familiares, características del EEG, de las neuroimágenes y resultados de otros estudios complementarios.
Los síndromes epilépticos pediátricos se presentan en el contexto de un cerebro que está teniendo marcados cambios en su estructura y funcionamiento, por lo tanto sus características clínicas y eléctricas se influencian fuertemente por modificadores edad-dependientes. En este contexto es posible observar sobreposición de características clínicas entre dos cuadros del mismo o incluso de otro grupo etario, lo cual agrega dificultad al reconocimiento certero del síndrome; también ocurre la evolución de un síndrome a otro en función de la edad o cambios en el perfil evolutivo de una epilepsia que por ejemplo, de ser refractaria, pasa a tener buena respuesta a fármacos antiepilépticos (FAEs). A su vez, la condición de activo cambio de la estructura cerebral, agrega vulnerabilidad a condiciones como las encefalopatías epilépticas, que devienen en deterioro cognitivo y neurológico permanentes.
EpidemiologíaLa epilepsia es una condición frecuente en todas las etapas de la niñez y adolescencia. Los estudios en niños en cuanto a su incidencia, aportan datos generalmente limitados a algunos grupos o situaciones específicas. La mayor incidencia, entendida como casos nuevos/año, ocurre en los primeros meses de vida, con cifras de hasta 130/100.000 (1), con una caída a 40/100.000 después del primer año, para permanecer relativamente estable durante la primera década de vida. En la edad adolescente la incidencia nuevamente disminuye a 20/100.000, cifra que se mantiene similar en la edad adulta. En países desarrollados, un 50% de las epilepsias se inician en la niñez o adolescencia. En cuanto a la incidencia de síndromes específicos, para síndrome de West es de 2-7/10.000 RN vivos (1, 2); Epilepsia focal benigna con espigas centro-temporales (BECTS) tiene una incidencia de 14-24% en las epilepsias de nuevo diagnóstico en menores de 15 años (3); la Epilepsia Mioclónica Juvenil (EMJ) y la Epilepsia de Ausencia corresponden cada una a cerca de un 1% del total de nuevos casos diagnosticados de epilepsia (4).
La prevalencia de epilepsias, entendida como los casos observados en un momento, es mayor que la incidencia y se estima entre 2,7-40/1000. Las cifras reportadas son menores en países desarrollados en comparación con estudios realizados en África, América Latina o Asia.
Clasificación de epilepsias y síndromes epilépticosClásicamente las epilepsias y síndromes epilépticos se han clasificado en función de dos ejes:
- a)
Sitio de origen de las crisis: generalizados/parciales.
- b)
Etiología: idiopáticos/criptogénicos/sintomáticos (6).
Aunque esta dicotomía tiene utilidad práctica, es ampliamente conocida y ha sido utilizada por los clínicos en forma extendida durante dos décadas, ha sido objeto de críticas que han llevado a una nueva propuesta en 2010 (7). En primer lugar se plantea que los términos generalizado/ parcial no significan lo mismo si hablamos de crisis o de síndromes; ejemplos de ello son los espasmos epilépticos aparentemente “generalizados” que se observan en el contexto de una epilepsia focal como ocurre en el Síndrome de West, o las crisis focales en el Síndrome de Dravet, producto de una mutación en el gen SCN1A.
En segundo lugar se propone una nueva clasificación etiológica, principalmente atendiendo a la explosión de avances genéticos y de neuroimágenes que han permitido precisar causas y fisiopatología de varios cuadros; se desincentiva el término “idiopático”, entendido como sinónimo de genético, benigno y con excelente respuesta a tratamiento farmacológico, a la luz de la descripción de un número progresivamente mayor de alteraciones genéticas en síndromes epilépticos cuya evolución dista de ser benigna; por otra parte algunos autores cuestionan el carácter de benigno o idiopático de algunos síndromes bien definidos como BECTS en que se ha descrito alteraciones del aprendizaje, lenguaje y conductuales y, evoluciones atípicas como el síndrome opercular; asimismo este cuadro se ha asociado a otros síndromes de pronóstico reservado, como Síndrome de Landau Kleffner y Estado Epiléptico durante el Sueño (ESES), planteándose que forman un continuo (8). El término “criptogénico”, definido como presumiblemente sintomático pero sin evidencias de ello, se deja de lado considerando que su aplicación es dependiente de la acuciosidad del estudio etiológico y de la disponibilidad de recursos tecnológicos. De este modo se propone clasificar las causas subyacentes a las Epilepsias en:
a) Genéticas: la epilepsia es el resultado directo de una alteración genética, en que las crisis epilépticas son una manifestación central; p.e. Síndrome de Dravet y gen SCN1A.
b) Estructurales-metabólicas: hay una condición estructural o metabólica que tiene un riesgo demostradamente mayor de desarrollar epilepsia; incluye lesiones adquiridas (AVE, trauma, infecciones), lesiones de origen genético (Esclerosis Tuberosa, alteraciones del desarrollo cortical) entendiendo que hay una lesión estructural entre el defecto genético y la epilepsia.
c) De causa desconocida.
Así la clasificación actualmente en uso reconoce las siguientes categorías:
-Síndromes electro-clínicos, ordenados por edad de inicio, se restringe a un grupo de entidades clínicas que se identifican de forma fiable por un conjunto de características electroclínicas.
-Constelaciones distintivas: representan constelaciones clínicamente diferenciadas sobre la base de lesiones específicas u otras causas; su reconocimiento tiene relevancia para el tratamiento.
-Epilepsias estructurales/metabólicas, secundarias a factores etiológicos definidos, pero que no se ajustan a un Síndrome electro-clínico específico.
-Epilepsias de causa desconocida. (Tabla 1).
Tabla 1.Síndromes electroclínicos y otras epilepsias
SÍNDROMES ELECTROCLÍNICOS ORGANIZADOS POR EDAD DE INICIO (A) EPILEPSIAS DE CAUSA DESCONOCIDA PERIODO NEONATAL - Condiciones con crisis epilépticas que no se diagnostican tradicionalmente como una forma de epilepsia per se - Epilepsia neonatal familiar beniga (BFNE) - Crisis neonatales benignas (BNS) - Encefalopatía mioclónica temprana (EME) - Crisis febriles (FS) - Síndrome de Ohtahara LACTANCIA - Epilepsia de la infancia (del lactante) con crisis focales migratorias CONSTELACIONES DISTINTIVAS - Síndrome de West - Epilepsia mioclónica de la infancia (del lactante) (MEI) - Epilepsia temporal mesial con esclerosis del hipocampo (MTLE with HS) - Epilepsia benigna de la infancia (del lactante) - Síndrome de Rasmussen - Epilepsia benigna de la infancia (del lactante) familiar - Crisis gelásticas con hamartoma hipotalámico - Síndrome de Dravet - Epilepsia hemiconvulsión-hemiplejia - Encefalopatía mioclónica en trastornos no progresivos - Las epilepsias que no encajen en ninguna de estas categorías diagnósticas se pueden distinguir primero por la presencia o ausencia de una condición conocida estructural o metabólica (supuesta causa) y luego por la forma primaria de inicio de las crisis (generalizadas vs focales) NIÑEZ - Crisis febriles plus (FS+) (puede empezar en la lactancia)
- Síndrome de Panayiotopoulos - Epilepsia con crisis mioclónico atónicas (previamente astáticas) - Epilepsia benigna con puntas centrotemporales (BECTS) - Epilepsia frontal nocturna autosómica dominante (ADNFLE) - Epilepsia occipital de la infancia de inicio tardío (tipo Gastaut) EPILEPSIAS ATRIBUÍDAS A Y ORGANIZADAS SEGÚN CAUSAS ESTRUCTURALES-METABÓLICAS - Epilepsia con ausencias mioclónicas - Síndrome de Lennox-Gastaut - Encefalopatía epiléptica con punta onda continua durante el sueño (CSWS) (b) - Malformaciones del desarrollo cortical (hemimegalencefalia, heterotopias, etc) - Síndrome de Landau-Kleffner (LKS)
- Síndromes neurocutáneos (complejo esclerosis tuberosa, Sturge-Weber, etc) - Epilepsia ausencia infantil (CAE) ADOLESCENCIA - EDAD ADULTA - Tumor - Epilepsia ausencia juvenil (JAE) - Infección - Epilepsia mioclónica juvenil (JME) - Trauma - Epilepsia con crisis generalizadas tónico-clónicas únicamente - Angioma - Epilepsias mioclónicas progresivas (PME) - Lesiones perinatales - Epilepsia autosómica dominante con características auditivas (ADEAF) - Accidentes cerebrovasculares etc… - Otras epilepsias familiares del lóbulo temporal - Relación menos específica con la edad - Epilepsia familiar focal con focos variables (infancia a edad adulta) - Epilepsias reflejas a) la disposición de los síndromes electroclínicos no refleja la etiología; b) algunas veces denominado Estatus Epilepticus durante el Sueño Lento (ESES)
Ref. (7).
El diagnóstico se ha basado en la evaluación clínica y principalmente en los resultados que entregan los exámenes de EEG y Neuroimágenes. Se enfatiza los siguientes elementos:
- •
Identificación del tipo de crisis y definir si su presencia debe ocurrir, puede ser parte o excluye el diagnóstico.
- •
Edad de inicio: en muchos síndromes hay rangos de edades de inicio que son distintivos, sin embargo en otros no hay consenso en cuan estrictamente aplicar este criterio.
- •
perfil progresivo: evidencia que sugiera que hay un proceso neurodegenerativo o de deterioro del neurodesarrollo que depende de la epilepsia y que afecta la evolución del síndrome, como ocurre en las Encefalopatías Epilépticas condiciones en las cuales el deterioro neurológico es consecuencia principalmente de la actividad epiléptica, a diferencia del deterioro secundario a una alteración metabólica, degenerativa o encefalítica subyacente.
- •
EEg interictal: determinar si los hallazgos del registro son condición necesaria, pueden observarse en algunos casos o excluyen el diagnóstico.
- •
Signos y síntomas interictales asociados: en particular aquellos derivados del examen neurológico y neuropsicológico, distinguiendo aquellos déficits atribuibles a la causa de la epilepsia p.e, hemiparesia en un paciente con quiste porencefálico, de aquellos secundarios a la farmacoterapia, o de los que genera la propia epilepsia, p.e. en una Encefalopatía Epiléptica.
- •
Mecanismos fisiopatológicos: alteraciones estructurales permanentes que pueden ser generalizadas o localizadas; enfermedades o alteraciones genéticas que causan epilepsia p.e. Síndrome de Angelman; mecanismos genéticos específicos que son propios de un síndrome y que lo diferencian de otros p.e. Síndrome de Dravet, Epilepsia frontal nocturna autosómica dominante (ADNFLE) (9).
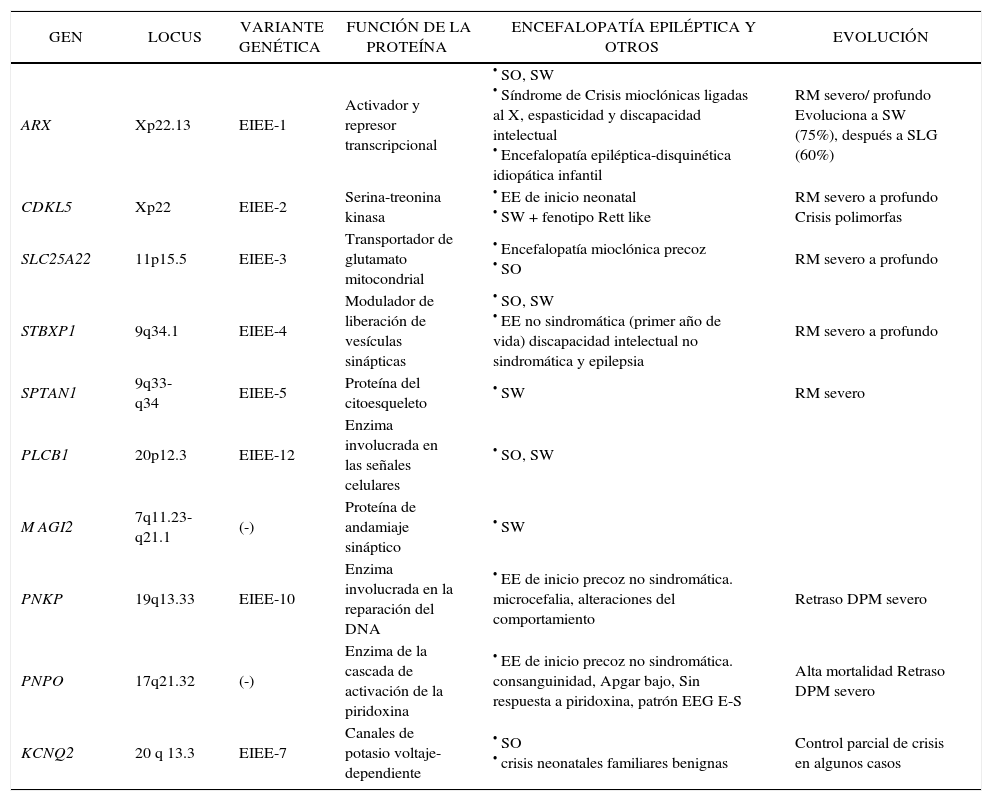
Un importante cuerpo de conocimiento generado desde la genética, que promete acrecentarse en los próximos años, ha alimentado el área de epilepsias pediátricas aportando a la comprensión de los mecanismos básicos que la subyacen. A los ya conocidos exámenes diagnósticos de uso clínico, que corroboran enfermedades malformativas, síndromes neurocutáneos o errores innatos del metabolismo que tienen como manifestación relevante la epilepsia, se agrega la investigación en epilepsias monogénicas, incluyendo epilepsias familiares (Epilepsia familiar neonatal benigna, GEFS+, ADNFLE), y Encefalopatías Epilépticas causadas por mutaciones de novo en genes específicos (Tabla 2). En el caso de las epilepsias familiares, ha sido elemento fundamental contar con familias numerosas con muchos miembros afectados. De otro lado, los avances en Encefalopatías Epilépticas ejemplifican la relevancia de tener una caracterización detallada del fenotipo clínico y EEG, tarea en que la escuela francesa ha sido pionera. El caso de Síndrome de Dravet es ejemplo de una entidad individualizada con criterios clínicos y EEG mucho antes que su mecanismo básico fuese identificado. La descripción clínica precisa y detallada permite el reconocimiento de nuevos síndromes -como Epilepsia de la Infancia (del lactante) con crisis focales migratorias- y desde allí orientar los estudios genéticos.
Genes asociados a encefalopatías epilépticas en niños menores
| GEN | LOCUS | VARIANTE GENÉTICA | FUNCIÓN DE LA PROTEÍNA | ENCEFALOPATÍA EPILÉPTICA Y OTROS | EVOLUCIÓN |
|---|---|---|---|---|---|
| ARX | Xp22.13 | EIEE-1 | Activador y represor transcripcional | • SO, SW • Síndrome de Crisis mioclónicas ligadas al X, espasticidad y discapacidad intelectual • Encefalopatía epiléptica-disquinética idiopática infantil | RM severo/ profundo Evoluciona a SW (75%), después a SLG (60%) |
| CDKL5 | Xp22 | EIEE-2 | Serina-treonina kinasa | • EE de inicio neonatal • SW + fenotipo Rett like | RM severo a profundo Crisis polimorfas |
| SLC25A22 | 11p15.5 | EIEE-3 | Transportador de glutamato mitocondrial | • Encefalopatía mioclónica precoz • SO | RM severo a profundo |
| STBXP1 | 9q34.1 | EIEE-4 | Modulador de liberación de vesículas sinápticas | • SO, SW • EE no sindromática (primer año de vida) discapacidad intelectual no sindromática y epilepsia | RM severo a profundo |
| SPTAN1 | 9q33-q34 | EIEE-5 | Proteína del citoesqueleto | • SW | RM severo |
| PLCB1 | 20p12.3 | EIEE-12 | Enzima involucrada en las señales celulares | • SO, SW | |
| M AGI2 | 7q11.23-q21.1 | (-) | Proteína de andamiaje sináptico | • SW | |
| PNKP | 19q13.33 | EIEE-10 | Enzima involucrada en la reparación del DNA | • EE de inicio precoz no sindromática. microcefalia, alteraciones del comportamiento | Retraso DPM severo |
| PNPO | 17q21.32 | (-) | Enzima de la cascada de activación de la piridoxina | • EE de inicio precoz no sindromática. consanguinidad, Apgar bajo, Sin respuesta a piridoxina, patrón EEG E-S | Alta mortalidad Retraso DPM severo |
| KCNQ2 | 20 q 13.3 | EIEE-7 | Canales de potasio voltaje-dependiente | • SO • crisis neonatales familiares benignas | Control parcial de crisis en algunos casos |
Adaptado de: Mastrangelo M, Leuzzi V. Genes of early-onset epileptic encephalopathies: from genotype to phenotype. Pediatr Neurol. 2012 Jan;46(1):24-31.
EE: encefalopatía epiléptica; EIEE: early infantile epileptic encephalopathies; SO: Síndrome de Ohtahara; SW: Síndrome de West; SLG: Síndrome de Lennox-Gastaut
Considerando que la data disponible es limitada, se estima que en general entre 65-75% de los niños que inician epilepsias, logran una remisión completa en el largo plazo, en tanto que 10–15% evolucionan a epilepsias refractarias (10, 11). Es interesante señalar que se estima en 20-40% de pacientes con epilepsias recién diagnosticadas que tienen remisiones espontáneas, sin tratamiento o con tratamientos con FAEs a mínimas dosis (12). La respuesta a tratamiento en niños (13), es a primera vista similar a lo observado en adultos (14), un 46% remite por más de un año con el primer FAE, 19% con el segundo y 9% con otros esquemas; sin embargo cabe hacer notar que en niños, un porcentaje mayor de los que fracasan con el primer fármaco, logran buena respuesta posterior (58% vs 32%). El mejor predictor de buen pronóstico se asocia a epilepsias “idiopáticas”, ya sean generalizadas o focales. Por otra parte el grupo de mal pronóstico se relaciona con una etiología sintomática, con tipo de crisis (espasmos, crisis tónicas, atónicas, parciales sintomáticas/criptogénicas) menor edad de inicio e historia de crisis febriles (CF).
A continuación se discuten algunos Síndromes Electroclínicos, incluidos en la Clasificación de ILAE (Tabla 1), atendiendo a criterios de frecuencia, relevancia de un diagnóstico preciso y aportes recientes de significación.
APeríodo NeonatalaEpilepsia Neonatal Familiar BenignaTambién denominadas crisis del tercer día, tienen una incidencia de 14.4/100 000 RN vivos, sin predominio de sexo. Se inician entre 2-5 días de vida en neonatos con historia familiar de crisis neonatales, sanos, sin factores precipitantes. Son crisis clónicas o tónicas breves, de 1–2 min, con frecuencia de 20-30/día, presentes durante el sueño o al despertar. El EEG interictal es normal o con anormalidad focal o multifocal. El EEG ictal muestra aplanamiento generalizado y sincrónico de 5-19s de duración, seguido por descarga focales o generalizadas de espigas, o punta-onda lenta.
La herencia es AD habiéndose identificado más de setenta mutaciones en el gen KCNQ2 y algunas en KCNQ3; ambos genes codifican para subunidades de canales de K voltaje dependiente. Los antecedentes familiares son característicos, la evaluación neurológica y los estudios complementarios son normales.
Generalmente remiten sin farmacoterapia específica. Es posible administrar benzodiazepinas, fenobarbital (PB) o fenitoína (PHT). Las crisis remiten entre 1-6m después del inicio. El pronóstico es generalmente bueno, un 11% presentará crisis febriles y 7% desarrolla problemas de aprendizaje. Excepcionalmente evolucionan a una encefalopatía epiléptica fármaco-resistente (15).
bSíndrome de ohtahara (SO) o encefalopatía epiléptica infantil precozEs la Encefalopatía Epiléptica edad-dependiente más precoz y menos frecuente. Debuta en los dos primeros meses de vida, en su mayoría antes del décimo día de vida. Las crisis corresponden a espasmos tónicos frecuentes, aislados o en salvas de hasta 10 segundos que ocurren tanto en sueño como en vigilia. En un tercio a la mitad de los casos se asocian a crisis focales motoras o a mioclonías masivas. El patrón EEG corresponde a estallido supresión (E-S) caracterizado por paroxismos de ondas lentas de alto voltaje, con espigas multifocales sobreimpuestas, que alternan con un trazado plano o de muy baja amplitud. En los casos secundarios a lesión o malformación, se observan asimetrías o alteraciones focales. No hay grafoelementos normales del sueño. El EEG ictal corresponde a atenuación difusa de la actividad de base con ritmos rápidos (RR) de bajo voltaje asociados (electrodecremento).
El examen neurológico es anormal con alteraciones del tono: hipotonía o espasticidad, frecuentemente asimétrica. La etiología es heterogénea, lo más frecuente son las alteraciones estructurales del SNC, luego enfermedades metabólicas: encefalopatía de Leigh y citopatías mitocondriales entre otras, y alteraciones genéticas por mutaciones en genes reguladores de funciones neuronales o interneuronales (Tabla 2).
Entre los 2-6 meses de vida el patrón de E-S disminuye, transformándose en hipsarritmia. Posteriormente a la edad de un año, evoluciona hacia espiga onda lenta difusa o espigas multifocales (SLG). La respuesta a farmacoterapia es decepcionante, ACTH y/o corticoides son a veces útiles. Se han utilizado altas dosis de vitamina B6 así como acido valproico (VPA), vigabatrina (VGB), hidrato de cloral o dieta cetogénica, con pobres resultados. Algunos pacientes se benefician del tratamiento quirúrgico. El pronóstico del SO es desfavorable, 8/16 pacientes reportados por Ohtahara fallecieron y el resto quedó con secuelas neurológicas importantes (16).
BPeríodo de LactanteaEpilepsia de la infancia (del lactante) con crisis focales migratoriasEs una encefalopatía epiléptica que debuta antes de los 6 meses y evoluciona hacia crisis focales subcontinuas que migran de una región a otra de la corteza cerebral, asociado a un importante deterioro del DPM. Se inicia entre la primera semana y los 7 meses de vida, con un promedio de 3 meses. Las crisis se caracterizan por manifestaciones motoras y autonómicas, con generalización secundaria en la mitad de los pacientes. Se describe desviación oculo-cefálica, sacudidas laterales de los ojos, mirada fija, sacudidas clónicas palpebrales, hipertonía o clonías de una extremidad o de un hemicuerpo, masticación, apneas, eritema facial, cianosis y salivación. El EEG interictal muestra un enlentecimiento basal, con asimetría fluctuante y puntas multifocales que no se activan en el sueño. Los husos de sueño son escasos y asimétricos. El EEG ictal se caracteriza por actividad theta rítmica que comienza en una región y se extiende progresivamente a regiones vecinas disminuyendo de frecuencia. Las crisis se superponen, una crisis comienza antes que finalice la anterior; la actividad ictal es continua y migrante, conformando estados epilépticos. La topografía de las descargas se correlacionan con la semiología clínica.
Al final del primer año de vida, las crisis se presentan en “clusters” de varias semanas de duración, asociándose a deterioro cognitivo. Las mioclonías y los espasmos epilépticos son infrecuentes. En el examen neurológico destaca hipotonía axial, sialorrea, trastorno de la deglución, ausencia de prehension voluntaria, contacto ocular inestable, la RM cerebral no muestra malformación cortical (17). Recientemente se han descrito mutaciones en el gen KCNT1 (19) y en el gen TBC1D24, esta última en dos hermanos afectados (20).
La evolución es hacia una microcefalia adquirida y epilepsia fármacoresistente; tanto FAEs como corticoides son ineficaces. Hay reportes de control de crisis con dosis altas de benzodiazepinas, asociadas a stiripentol, o con bromuros. Algunos niños mueren antes del primer año de vida, por crisis particularmente frecuentes y complicaciones respiratorias.
bSíndrome de WestDescrito por West en 1841 (20), se define por la tríada de espasmos infantiles, retraso/deterioro del DPM e hipsarritmia (21). Las crisis de espasmos epilépticos son contracciones axiales breves y bruscas, que duran entre 0.2 a 2 segundos, más frecuentes en flexión que en extensión, que se repiten en salvas, separados por 5-20 seg. Pueden acompañarse de llanto o limitarse a una contracción del cuello o elevación de los ojos. Las asimetrías o elementos focales en los movimientos de ojos, cabeza o extremidades, automatismos o nistagmus, orientan hacia una lesión cerebral. La frecuencia diaria de crisis es alta, sobre todo en transiciones sueño-vigilia. Puede haber crisis focales, previas o conjuntas a los espasmos. El desarrollo psicomotor (DPM) inicial puede ser normal o anormal según la etiología, luego ocurre una regresión global, que no se correlaciona con la frecuencia de los espasmos, pero sí con las anomalías interictales.
Encefalopatías epilépticas: características clínicas y eeg de síndromes de ohtahara, west y lennox-gastaut
| SINDROME OHTAHARA | SINDROME WEST | SINDROME LENNOX-GASTAUT | |
|---|---|---|---|
| Descripción | 1976 por Ohtahara | 1841 por West | 1950 por Gastaut |
| Edad de inicio | 0-3 meses | 4-8 meses | 2-7 años |
| Tipo de crisis | Espasmos tónicos Crisis focales | Espasmos masivos Crisis focales (25 % de los casos) | Crisis tónicas (1) Ausencias atípicas (2) Crisis atónicas (3) |
| EEg interictal | Estallido- supresión | Hipsarritmia | Complejos espiga-onda lenta |
| EEg ictal | E-S o electrodecremento | Electrodecremento | Actividad rápida de bajo voltaje, de frecuencia y amplitud creciente complejos espiga-onda lenta polipuntas ondas o punta onda generalizadas |
| genes asociados | ARX-CDKL5-SLC25A22 - STXBP1- KCNQ2 | ARX-CDKL5-SLC25A22 - STXBP1-SPTAN1 PLCbeta1, MAGI2 y PNKP | |
| pronóstico | Desfavorable | Desfavorable | Desfavorable |
El EEG interictal típico es la hipsarritmia caracterizada por una desorganización completa del trazado de base, con ondas lentas de alto voltaje, bilaterales y simétricas, con espigas multifocales irregulares. Este aspecto es continuo tanto en sueño como en vigilia; en sueño las espigas tienden a sincronizar dando el aspecto de un trazado fragmentado. En 33% de los pacientes se describen hipsarritmias atípicas o modificadas. El EEG ictal muestra una onda lenta de gran amplitud seguida de atenuación de voltaje (electrodecremento), asociada a ritmos rápidos o en ocasiones, sólo actividad rápida de bajo voltaje.
Un 75% de los SW son sintomáticos, entre 7-13% son secundarios a Esclerosis Tuberosa. El SW idiopático es poco frecuente (5%), se caracteriza por un DPM previo normal, espasmos simétricos e hipsarritmia típica. La etiología es el principal factor para el pronóstico cognitivo y de la epilepsia (Tabla 4). En el corto plazo el 50% de los casos sintomáticos y el 80% de los criptogénicos, responde clínicamente desde el inicio del tratamiento (ACTH o VGB), desapareciendo la hipsarritmia (22). A los 3 años de seguimiento, solo un 16% de los niños con SW tienen un desarrollo normal (23). En el largo plazo entre 11-17% tienen inteligencia normal, observándose una alta mortalidad de hasta un 30%, concentrada en los tres primeros años de vida (24). Aportan a un mejor pronóstico: DPM normal previo, regresión psicomotora poco importante, ausencia de causa reconocible, ausencia de signos neurológicos focales y de otro tipo de crisis, tratamiento precoz y respuesta rápida al tratamiento. Un 12 % de los SW evolucionan a Síndrome de Lennox-Gastaut (SLG).
Etiología de síndrome de west
| ETIOLOGIA | CAUSAS | |
|---|---|---|
| Sintomática 75% | Malformaciones cerebrales 30% | Agenesia del cuerpo calloso, polimicrogiria, agiria-paquigiria, lisencefalia, hemimegaencefalia, displasias corticales focales, esquizencefalia |
| Síndrome neurocutáneos | Esclerosis tuberosa (7-13%), Neurofibromatosis, Incontinencia Pigmenti, Hipomelanosis de Ito | |
| Cromosómicas | Síndrome Down, XXY, 22q, 17p13.3del, Inv/dupl15, 1p36del, del1q36 1ptel, | |
| Genéticas | ARX-CDKL5-SLC25A22-STXBP1-SPTAN1-PLCbeta1, MAGI2 y PNKP | |
| Encefalopatía hipóxico isquémica | Asfixia perinatal o asfixia por inmersión | |
| Errores innatos del metabolismo | Enfermedad de Menkes, PKU, déficit de tetrahidrobiopterina, acidurias orgánicas, enfermedades mitocondriales, déficit de piridoxina | |
| Infecciones pre, peri y postnatales | Meningitis, CMV, herpes, toxoplasmosis | |
| AV E | ||
| Traumatismos | Trauma de parto | |
| Criptogénicas 9-15 % | ||
| Idiopáticas 5-10 % |
En el tratamiento se ha demostrado la utilidad de corticoides, prednisolona, ACTH natural y sintética, hidrocortisona, metilprednisolona, existiendo variedad de esquemas de dosis y duración. ACTH es más eficaz que prednisona y una mayor dosis se relaciona con mejor respuesta. Los efectos secundarios son el mayor inconveniente del uso de corticoides. VGB es más eficaz en pacientes que inician tratamiento antes de los 3 meses de vida, con mejor eficacia en Esclerosis Tuberosa y en displasias corticales focales. Su mayor problema es el riesgo de disminución del campo visual que depende de las dosis acumulativas. Otros FAEs que han reportado eficacia son VPA, clobazam, nitrazepam y altas dosis de piridoxina. Con los nuevos FAEs como topiramato (TPM), felbamato, zonisamida, levetiracetam (LEV), la tasa de respuesta es baja. La dieta cetogénica ha mostrado utilidad en algunos casos.
cSíndrome de Dravet (SD) o Epilepsia Mioclónica Severa del lactantedescrita en 1978, es una entitad epiléptica compleja caracterizada por crisis polimorfas y resistentes a tratamiento, asociadas a la instalación progresiva de trastornos cognitivos y de la personalidad. Su incidencia es de 1/30.000-40.000 y representa el 5% de las epilepsias que debutan en el primer año de vida. En la fase inicial hay crisis clónicas generalizadas o hemigeneralizadas, subfebriles, prolongadas, que pueden evolucionar a estado epiléptico. También pueden asociarse a crisis reflejas por agua caliente. En esta fase tanto DPM como examen neurológico y neuroimágenes son normales. El EEG es habitualmente normal, sin embargo algunos pacientes pueden tener fotosensibilidad y un enlentecimiento theta en las regiones centro-parietales. En la segunda fase entre 1-6 años, los estados epilépticos son menos frecuentes, aparecen crisis mioclónicas y se observan además ausencias atípicas con un componente mioclónico frecuente. En un 40-50 % de los casos se presentan crisis focales complejas, de semiología variable y sin valor localizatorio. Pueden persistir las crisis generalizadas clónicas o tónico-clónicas (TCG) en un contexto subfebril o afebril, nocturnas o diurnas y ser provocadas por el contacto con agua, incluso con agua de mar, o por la actividad física intensa. El EEG de base se hace más lento y desorganizado. Se registran descargas de punta-onda generalizadas que aparecen sobre todo durante el sueño, asociadas a anomalías focales y multifocales; la fotosensibilidad se observa en <10%. En esta etapa, se hace evidente un trastorno del lenguaje y ataxia asociada a reflejos vivos. El contacto ocular es difícil, asociado a hiperactividad, inatención, distractibilidad, excesiva familiaridad y/o impulsividad.
En el largo plazo hay una tendencia a la estabilización, con disminución de la frecuencia de crisis. Persisten las CTCG o focales, más raramente tónicas nocturnas o diurnas. Se logra una adquisición parcial del lenguaje y la marcha es atáxica. La evolución cognitiva es hacia un retardo mental de severidad variable. Cerca de un 15% de los pacientes presenta muerte súbita durante el sueño, cuyo mecanismo se desconoce, pudiendo producirse en el transcurso de una crisis en el sueño o por alteraciones del ritmo cardíaco (25).
Genética: El SD es esporádico, en 25% hay historia familiar de epilepsia. Se han documentado diferentes mutaciones en el gen SCN1A, que codifica para una subunidad del canal de sodio; estas mutaciones, de novo en el 95%, se encuentran en más del 70% de los pacientes; no hay correlación genotípica-fenotípica. Las mutaciones SCN1A producen disminución de función de los canales de sodio voltaje-dependientes tipo 1, sobre todo en las interneuronas gabaérgicas. Es por esto que lamotrigina (LTG), que bloquea los canales de sodio, no es eficaz sobre la epilepsia; por otra parte, sí tendrían utilidad las benzodiazepinas, que aumentarían una transmisión gabaérgica deficitaria.
El estudio de mutaciones del gen SCN1A debe considerarse en todas las personas con un posible SD, con la clínica inicial típica de un lactante normal que presenta crisis febriles o afebriles, recurrentes, prolongadas, hemiclónicas o un estado epiléptico generalizado; después de los dos años, con la presencia de otros tipos de crisis y detención del desarrollo, el diagnóstico clínico se hace más claro. En mujeres, cuando no se encuentra mutaciones en el estudio de SCN1A, deberá plantearse la “Epilepsia y RM limitados a las mujeres”, causado por mutaciones en la protocaderina 19 (PCDH19), con una herencia ligada al X inusual, que se expresa solamente en mujeres. También se han registrados mutaciones en el gen de la subunidad alfa 2 del receptor GABA A (GABRG2), así como una mutación homocigota en el gen SCN1B (26).
El tratamiento de SD es difícil, la asociación de VPA, stiripentol y clobazam es eficaz en disminuir la frecuencia de los estados epilépticos. Otros fármacos que han mostrado cierta eficacia son TPM, LEV y PB. Por su mecanismo de acción, los fármacos que pueden agravar las crisis son: carbamazepina (CBZ), oxcarbazepina (OXC), LTG, PB y PHT.
CNiñezaEpilepsia con Crisis febriles plus (GEFS+)Descrita en 1997 por Scheffer y Berkovic, es un síndrome epiléptico genéticamente determinado que incluye crisis febriles típicas y epilepsia generalizada que se presentan en la infancia. Las crisis febriles (CF) plus se distinguen de las crisis simples o complejas por su persistencia más allá de la edad de 6 años o su asociación a crisis afebriles. La edad de inicio es variable ya sea en la infancia, sin intervalo libre en relación a las C F, ya sea en la edad adulta. Pueden estar asociadas a otros tipos de crisis: ausencias, mioclonías y crisis atónicas, o también estar asociadas a un síndrome más severo como Epilepsia con crisis mioclónico-atónicas (antes astáticas) (EMA) (27).
Los estudios genéticos de grandes familias con GEFS+ demuestran una herencia AD con penetrancia variable. Se han identificado mutaciones en los genes SNC1A (más frecuente), SCN1B, GABRG2 (que codifica para la subunidad gamma 2 del receptor GABA A), SCN2A que es el mismo canal de sodio voltaje-dependiente pero en la subunidad alfa 2 y del gen GABRD, que codifica para la subunidad delta del receptor GABA A. Algunos estudios genéticos señalan que GEFS+ pertenece a un espectro clínico, que también incluye al SD que estaría en el extremo más severo. El examen neurológico y neuroimágenes son normales. El EEG presenta actividad epileptiforme generalizada en la epilepsia activa.
bSíndrome de Panayiotopoulos o Epilepsia Occipital de Inicio TempranoPertenece a las epilepsias focales benignas, es un cuadro frecuente, edad-dependiente y con susceptibilidad genética. Las crisis se inician a los 5 años (3-6 a), son poco frecuentes, prolongadas de más de 30 min de duración, y con fenómenos autonómicos (vómitos, diaforesis, palidez, irritabilidad, síncope); pueden acompañarse de desviación ocular, alteración de la conciencia y evolucionar a una generalización. El EEG interictal tiene una actividad basal normal, con espigas occipitales o multifocales. En el EEG ictal se observa actividad rítmica theta de predomino posterior seguida de punta-onda lenta rítmica de amplitud alta en regiones occipitales, con generalización secundaria. Tanto examen neurológico como estudios complementarios son normales (28).
El diagnóstico diferencial es amplio e incluye encefalitis, migraña atípica, síncope cardiogénico, gastroenteritis. Si el evento es único no se recomienda el uso de FAEs dada la baja probablidad de recurrencias. Si las crisis son recurrentes se recomienda CBZ o OXC y como segunda opción clobazam, LEV, LTG o VPA. La evolución y el pronóstico son invariablemente buenos (29).
cSíndrome de Doose o Epilepsia con crisis Mioclónico-Atónicas (antes Astáticas) (EMA)Descrita por Doose en 1970, comparte con el SLG la edad de inicio, crisis generalizadas asociadas a veces a caídas, retraso del desarrollo y en el EEG, complejos punta-onda generalizados. A diferencia del SLG, no hay evidencia de lesión cerebral, los niños son previamente normales, con antecedentes de crisis febriles en 11-28% y presentan antecedentes familiares de epilepsia en 15-32%, sospechándose una etiología genética. Las crisis son de múltiples tipos: mioclónico-astáticas, ausencias, TCG y eventualmente crisis tónicas que le otorgan un pronóstico desfavorable. La edad de inicio es entre 18-60m, con un pic a los 3 años (30). Existen casos de EMA en familias con GEFS+, con mutaciones en los genes que codifican para canales de sodio (SNC1A y SNC1B) y para receptores GABA (GABRG2).
Al inicio el EEG tiene actividad basal normal o lentitud theta a 4-7 Hz, monomorfa, difusa, de predominio sobre regiones centro-parietales. Se asocian descargas breves de polipunta o punta-onda generalizadas, asimétricas a 2-3 Hz. En sueño hay mioclonías proximales asociadas a descargas generalizadas; el EMG muestra un paroxismo de 100 mseg, seguido por silencio post-mioclónico de 200-500 mseg; este último sería responsable de las caídas típicas de este sindrome. Clínicamente, el deterioro cognitivo combina dispraxia y disartria. En la minoría de los casos la evolución es desfavorable con la aparición de un estado epiléptico mioclónico, que se acompaña de una regresión del desarrollo, sobre todo del lenguaje, y de las funciones motoras, con ataxia, hipotonía y sialorrea. Las neuroimágenes no muestran anormalidades específicas.
La evolución de este síndrome es variable, un 50-89% deja de tener crisis al cabo de 3 años, un 58% con un CI normal. El tratamiento se mantiene a nivel empírico, se ha reportado éxito con VPA, etosuximida (ESM) y benzodiazepinas, en especial clobazam; la mejor combinación sería VPA y LTG, adjuntando ESM cuando predominan las mioclonías y las ausencias. La corticoterapia tiene indicación frente a un estado epiléptico mioclónico. PHT CBZ y VGB pueden empeorar las crisis. La dieta cetogénina se indica cuando hay evolución hacia una encefalopatía epiléptica (31).
dEpilepsia Benigna con Puntas Centrotemporales (BECTS) o Epilepsia RolándicaEs la epilepsia más frecuente en la edad escolar, dando cuenta de un 15% de ellas. De inicio entre los 3–14 años (75% entre 5–10a), desaparece en la segunda década de la vida. Afecta a niños previamente sanos, más frecuente en varones, con o sin historia familiar de BECTS. Las crisis son breves, focales motoras, hemifaciales, con síntomas somatosensoriales, con preservación de la conciencia, a veces con arresto del lenguaje y salivación abundante, generalmente fáciles de controlar y con tendencia a desaparecer. Un 75% ocurre durante el sueño y/o al despertar y el 10% en vigilia. El EEG interictal tiene una actividad basal normal, hay espigas o puntas de alto voltaje seguidas de onda lenta, unifocales o multifocales, centro-temporales, temporales medias, centro-parietales, fronto-centrales o centrooccipitales, que se activan con el sueño. Con tendencia a cambiar o propagarse de un hemisferio a otro. El EEG ictal presenta descargas de inicio centro-temporal unilateral con generalización hemisférica, seguido por propagación a regiones homólogas contralaterales (32). Hay alta predisposición genética, autosómica dominante (AD), con penetrancia edad-relacionada.
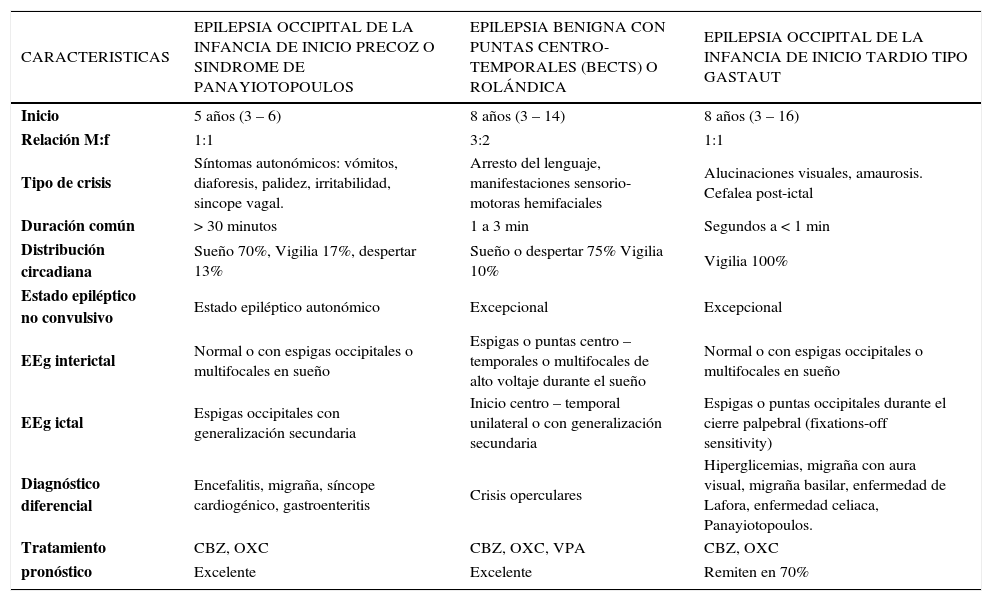
Características clínicas de las epilepsias focales benignas de la infancia
| CARACTERISTICAS | EPILEPSIA OCCIPITAL DE LA INFANCIA DE INICIO PRECOZ O SINDROME DE PANAYIOTOPOULOS | EPILEPSIA BENIGNA CON PUNTAS CENTRO-TEMPORALES (BECTS) O ROLÁNDICA | EPILEPSIA OCCIPITAL DE LA INFANCIA DE INICIO TARDIO TIPO GASTAUT |
|---|---|---|---|
| Inicio | 5 años (3 – 6) | 8 años (3 – 14) | 8 años (3 – 16) |
| Relación M:f | 1:1 | 3:2 | 1:1 |
| Tipo de crisis | Síntomas autonómicos: vómitos, diaforesis, palidez, irritabilidad, sincope vagal. | Arresto del lenguaje, manifestaciones sensorio-motoras hemifaciales | Alucinaciones visuales, amaurosis. Cefalea post-ictal |
| Duración común | > 30 minutos | 1 a 3 min | Segundos a < 1 min |
| Distribución circadiana | Sueño 70%, Vigilia 17%, despertar 13% | Sueño o despertar 75% Vigilia 10% | Vigilia 100% |
| Estado epiléptico no convulsivo | Estado epiléptico autonómico | Excepcional | Excepcional |
| EEg interictal | Normal o con espigas occipitales o multifocales en sueño | Espigas o puntas centro – temporales o multifocales de alto voltaje durante el sueño | Normal o con espigas occipitales o multifocales en sueño |
| EEg ictal | Espigas occipitales con generalización secundaria | Inicio centro – temporal unilateral o con generalización secundaria | Espigas o puntas occipitales durante el cierre palpebral (fixations-off sensitivity) |
| Diagnóstico diferencial | Encefalitis, migraña, síncope cardiogénico, gastroenteritis | Crisis operculares | Hiperglicemias, migraña con aura visual, migraña basilar, enfermedad de Lafora, enfermedad celiaca, Panayiotopoulos. |
| Tratamiento | CBZ, OXC | CBZ, OXC, VPA | CBZ, OXC |
| pronóstico | Excelente | Excelente | Remiten en 70% |
En su tratamiento son útiles CBZ, OXC y VPA. También puede usarse clobazam en dosis nocturna. Tratados o no, las características clínicas y EEG remiten antes de los 15-16 años en la gran mayoría de los casos. El pronóstico neurológico, cognitivo y de crisis en el largo plazo es bueno, aunque se han descrito algunas dificultades conductuales y en el aprendizaje escolar. Un número no determinado de casos presenta durante su evolución dificultades de lenguaje, aprendizaje y conducta, dificultades cognitivas transitorias en relación a mayor actividad paroxística en el EEG y activación de las descargas de punta onda en el sueño; estos casos se denominan BECTS plus. Otro grupo presenta evoluciones atípicas tales como status de BECTS o síndrome opercular adquirido. Se ha planteado que BECTS sería el extremo benigno de un espectro cuya expresión más severa sería ESES con deterioro marcado o regresión epileptiforme desintegrativa (33).
eEpilepsia Occipital de la infancia de inicio tardío (Tipo Gastaut)Se trata de un síndrome de inicio a los 8 años (3–16 años), probablemente determinada genéticamente, representa el 4% de todas las epilepsias benignas y el 0,38% del total de las epilepsias en edad pediátrica. Las crisis son frecuentes, diurnas y con componentes visuales: alucinaciones, amaurosis o ambos; pueden acompañarse de desviación ocular y/o generalización secundaria, en un 25% hay cefalea postictal. El EEG interictal muestra paroxismos de espiga-onda o punta-onda de amplitud alta, que recurren rítmicamente sobre regiones occipitales o temporales posteriores ya sea uni o bilateralmente, pero sólo con los ojos cerrados (fixation-off sensitivity). Durante las crisis la descarga puede propagarse hacia regiones centrales o temporales y generalizar (34). Entre los diagnósticos diferenciales están hiperglicemia, migraña con aura visual, migraña basilar, enfermedad de Lafora, calcificaciones occipitales en relación con la enfermedad celiaca, síndrome de Panayiotopoulos.
En su tratamiento son de utilidad CBZ, OXC, y como segunda línea, LEV, LTG o VPA; también es posible utilizar clobazam en dosis nocturnas (29).
fEpilepsia con Ausencias MioclónicasDescrita por Tassinari en 1971, fue reconocida como síndrome en 1989. Se inicia entre los 11m y 12a (promedio 7 años), con crisis de ausencia con mioclonías rítmicas bilaterales y difusas de intensidad severa de10-60 seg de duración, frecuentes y diarias; un 45% presenta crisis TCG. El EEG interictal revela una actividad de base normal y en 1/3 de los casos, complejos punta-onda generalizados. Durante los episodios de ausencia mioclónica hay descargas rítmicas de punta o poliespiga-onda lenta a 3 Hz, generalizadas, sincrónicas y simétricas.
El examen neurológico y las neuroimágenes son normales. El tratamiento de elección es VPA combinado con ESM, PB o clobazam. Las crisis de ausencia mioclónica remiten en alrededor de un 37% de los casos; en el resto persisten o evolucionan a otras formas de epilepsia. La presencia de crisis TCG se asocia a mal pronóstico (35).
gSíndrome de Lennox-GastautSe presenta antes del segundo año de vida, cuando hay un SW u otra epilepsia previa. Se caracteriza por múltiples tipos de crisis, déficit o regresión cognitiva y un patrón EEG típico. Las crisis son tónicas axiales, atónicas y ausencias atípicas; pueden también presentar crisis mioclónicas, generalizadas tónico-clónicas (CGTC), focales o espasmos. Las crisis tónicas son axiales, en extensión, acompañadas de revulsión ocular y pausa respiratoria, son más frecuentes durante la somnolencia. Las ausencias atípicas se presentan como un cese de la actividad e hipotonía axial, asociadas a complejos punta-onda lenta bilaterales entre 1,5 a 2,5 Hz, sincrónicos pero irregulares en frecuencia y amplitud. La frecuencia de crisis es elevada y es frecuente observar estados epilépticos generalizados de hasta varios días, que se manifiestan desde una simple obnubilación hasta un estado tónico.
El EEG interictal se caracteriza por una actividad de base anormal de punta onda lenta a <3 Hz tanto en sueño como en vigilia; durante el sueño aparecen paroxismos de ritmos rápidos, alrededor de 10 Hz y poliespiga-onda difusas.
La etiología es frecuentemente sintomática, en especial malformativa. Los casos calificados de idiopáticos corresponderían a otros síndromes, como Síndrome de Doose.
El tratamiento considera VPA, LTG), benzodiazepinas, VGB, Zonisamida, TPM, felbamato y rufinamida. La corticoterapia tiene utilidad cuando se administra precozmente. Las benzodiazepinas deben administrarse con precaución debido a que pueden desencadenar crisis tónicas. El estado epiléptico generalizado tónico se maneja con PHT. Las formas particularmente fármaco-resistentes se benefician con dieta cetogénica, estimulador vagal o cirugía (callosotomía o resección focal)
El pronóstico es oscuro, la epilepsia es severa y resistente a tratamiento; no hay mejoría del estado cognitivo (36).
hEncefalopatía epiléptica con punta onda continua durante el sueño o Status Epilepticus durante el Sueño Lento (ESES)Esta encefalopatía infrecuente, limitada en el tiempo, edad dependiente, debuta entre los 2-8 años, con predominio en varones. Se caracteriza por crisis focales o generalizadas, deterioro cognitivo y/o motor, con ataxia, dispraxia, distonía o déficit unilateral y EEG con punta-onda difusa, focal o unilateral que ocupa más del 85% del sueño lento y que persiste en tres registros realizados al menos en un mes. El deterioro cognitivo sin embargo es observable con un índice de ocupación de sueño de 50% o menos.
ESES se presenta en varias condiciones: evoluciones atípicas de BECTS, con mioclonus negativo o dispraxia facial; epilepsias sintomáticas que involucran región rolándica y tálamo, epilepsias criptogénicas que comprometen el lóbulo temporal con afasia y agnosia auditiva. Las crisis pueden presentarse antes del inicio del ESES; se han descrito crisis focales motoras, ausencias atípicas con componente atónico o tónico que llevan a caídas, crisis TCG o crisis focales complejas, así como también mioclonías negativas frecuentes en vigilia. En la mayoría de los casos las crisis desaparecen en el plazo de 4-14 años; en 30% desaparecen simultáneamente con la desaparición del ESES. El 50% de los casos queda con un CI inferior al inicial y con trastornos conductuales persistentes. El pronóstico se relacionaría con la duración del ESES.
El tratamiento debe ser precoz y activo. Las crisis se controlan con relativa facilidad, sin embargo las alteraciones en el EEG son difíciles de tratar. Se ha observado mejoría en EEG y en el estado neuropsicológico con benzodiazepinas, ACTH y corticodes. La corticoterapia debe ser prolongada para evitar recaídas. VPA, CBZ y LTG agravarían el cuadro (37).
Se ha descrito una asociación de ESES con crisis rolándicas, apraxia del habla y déficit cognitivo relacionados a una mutación del gen SRPX2 (38).
iEpilepsia Ausencia de la Niñez o PicnolepsiaEs una epilepsia generalizada primaria que corresponde a 10-15% de las epilepsias en edad escolar, se inicia entre los 3-13a con un pic a los 6-7a, más frecuente en niñas. Se presenta en pacientes previamente sanos. Las crisis corresponden a ausencias típicas, son frecuentes, caracterizadas por desconexión de segundos de duración sin pérdida de la postura, mirada fija, a veces asociadas a parpadeo, chupeteo, automatismos de manos, de término abrupto, sin postictal. Se desencadenan típicamente con la hiperventilación. Un 30-50% de los casos presentan crisis TCG. El trazado EEG interictal muestra una actividad basal normal y presencia de ondas delta rítmicas intermitentes occipitales (OIRDAS). Durante las crisis hay descarga de espiga–onda generalizada, sincrónicas y simétricas a 3 Hz. Las crisis pueden gatillarse por la hiperventilación (39).
Existe una predisposición genética elevada. entre 15-44% tiene historia familiar de epilepsias generalizadas idiopáticas y se reporta un 75% de concordancia en gemelos monocigotos. Se ha propuesto relación con genes que codifican para subunidades de canales de Ca, en especial CACNA1G y CACNA1H. Otro posible locus de susceptibilidad en subgrupos de pacientes es el gen CLCN2, gen que codifica para canales de Cl. Se ha descrito déficit de GLUT1 en pacientes con fenotipos de inicio precoz.
La primera línea de tratamiento es VPA y ESM; en segunda línea están LTG y LEV. Cuando es necesario, es posible usar asociaciones de VPA+LMT o VPA+ESM. Son potenciales agravantes de las crisis: CBZ, OXC, PB, PHT, VGB (40).
El pronóstico es generalmente bueno, las crisis ceden en la adolescencia, se reporta remisión en 56-84% de los casos. Cuando el inicio de las crisis es más temprano el pronóstico es excelente (41).
DAdolescenciaaEpilepsia de Ausencia JuvenilEpilepsia generalizada primaria que se inicia entre los 5-20a, con 70% de los casos entre 10-13a. Las crisis de ausencia típica son predominantes, menos frecuentes que en ausencias de la niñez (1-10/día); hay crisis TCG al despertar (80%) o crisis miclónicas (20%). El EEG interictal es normal. La actividad ictal comprende descargas de punta–onda lenta generalizada con acentuación frontal >3 Hz, desencadenadas por la hiperventilación y la fotoestimulación.
La exploración neurológica exámenes complementarios y RM cerebral son normales.
El tratamiento varía en función del sexo; en varones VPA como primera línea, LTG como segunda línea; en mujeres: LTG o VPA (no más de 1g/día) como primera línea, asociación LTG+VPA o ESM+LTG o PB+VPA.
No existe información precisa en torno al pronóstico; se estima que entre 37-62% de los pacientes entran en remisión y otro grupo debe tratarse de por vida. Un subgrupo evoluciona a Epilepsia Mioclónica Juvenil (41).
bEpilepsia Mioclónica Juvenil o Síndrome de JanzCorresponde a 5-10% de todas las epilepsias, con una incidencia de 1/1000-2000 habitantes. Se presenta en adolescentes y adultos previamente sanos (6-22a), 78% en la adolescencia, ligeramente más frecuente en mujeres que en hombres, sin diferencias raciales. Se caracteriza por crisis mioclónicas (100%), generalizadas, bruscas de predominio en hombros y manos, aisladas o en salvas, frecuentes al despertar; crisis TCG (80-90%) al despertar, bajo privación de sueño, alcohol, estrés o menstruación; ausencias (25-30%). El EEG interictal es normal. El EEG ictal muestra descargas rítmicas de poliespiga-onda lenta de >3 Hz generalizadas, sincrónicas y simétricas, poliespiga– onda tras el cierre palpebral (fixation-off sensitivity). La respuesta fotoparoxistica es anormal en 40-50% de los casos (42).
Un tercio de los pacientes tiene historia familiar de epilepsia. El fenotipo epiléptico varía entre familias y se han descrito las siguientes mutaciones: CACNB4 (mutación en la subunidad 4-beta del canal de Ca), GABRa1, GABRD, CLCN2, Myoclonin EFHC1 (43)
El examen neurológico y las neuroimágenes son generalmente normales.
Entre 80-90% de los pacientes en tratamiento logra libertad de crisis, sin embargo suelen requerir tratamiento antiepiléptico de por vida. En varones se indica VPA, LTG (eficaz en crisis de ausencias) o LEV (eficaz en crisis mioclónicas y en crisis fotosensibles). En mujeres LTG es la primera opción, seguida de VPA de liberación prolongada en dosis no >1 g/día o LEV. Las crisis pueden agravarse con CBZ, PHT o VGB (41).
ConclusiónLas epilepsias en la edad pediátrica son frecuentes y con manifestaciones clínicas diversas y edad-dependientes. Su reconocimiento preciso es importante para determinar un tratamiento y pronóstico adecuados. El mayor porcentaje (~70%) de los casos tiene una buena respuesta a tratamiento, remisión espontánea y buen pronóstico en el largo plazo. Un grupo menor, pero no menos importante, es refractario a tratamiento médico, destacando aquí las Encefalopatías Epilépticas que conllevan un mal pronóstico neurológico y un grupo de pacientes refractarios que serán candidatos a otras alternativas de tratamiento, como cirugía de la Epilepsia.
Las autoras declaran no tener conflictos de interés, con relación a este artículo.