






Ha habido debates importantes sobre bioequivalencia y sustitución de ciertos medicamentos de índice terapéutico estrecho, tales como los fármacos antiepilépticos. En este artículo revisamos los conceptos por detrás del tratamiento de la epilepsia, haciendo hincapié en las características específicas de estos medicamentos, así como los datos relacionados con las crisis epilépticas y el umbral epiléptico individual. Estas condiciones pueden volver vulnerables a los pacientes con epilepsia cuando se cambia sus fórmulas farmacéuticas, ya sea entre las de marca a los productos genéricos o similares y viceversa, así como entre los genéricos entre sí.
There has been considerable debate about bioequivalence and generic substitution of certain narrow therapeutic index drugs such as the antiepileptic drugs. Here we review concepts behind epilepsy treatment emphasizing specific characteristics of these medications as well as data related to seizures and individual epileptic threshold. These conditions may make patients with epilepsy vulnerable when switching of formulations is conducted, either between brand to generic or similar products and vice versa as well as between generics each other.
El Medicamento genérico es aquel similar a un producto de referencia, innovador o de marca, con el cual pretende ser intercambiable. Generalmente producido después de la expiración o renuncia de la protección de patentes u otros derechos de propiedad, es designado con el nombre científico del fármaco. El medicamento de referencia, innovador o de marca contiene una entidad química (fármaco) originalmente investigado y desarrollado por el laboratorio de fabricación, que es responsable de los estudios pre-clínicos y clínicos en los cuales se demostró la eficacia y la seguridad para las indicaciones y condiciones clínicas. Para la aceptación de un fármaco genérico, las autoridades reguladoras requieren la prueba de su eficacia, seguridad y calidad, así como pruebas de su bioequivalencia con el medicamento de referencia. Producto farmacéutico similar es el producto patentado que contiene los mismos ingredientes activos del producto de referencia, con la misma concentración, forma farmacéutica, vía de administración, dosis e indicación terapéutica, siendo equivalente al fármaco registrado en la agencia federal responsable de la vigilancia sanitaria. Estos fármacos sólo se diferencian de los productos de marca en las características relacionadas con el tamaño y la forma de las partículas, la vida útil, el envasado, el etiquetado, excipientes y vehículos. Para los medicamentos similares es obligatorio el uso de un nombre comercial, estando prohibido el uso del nombre genérico (1).
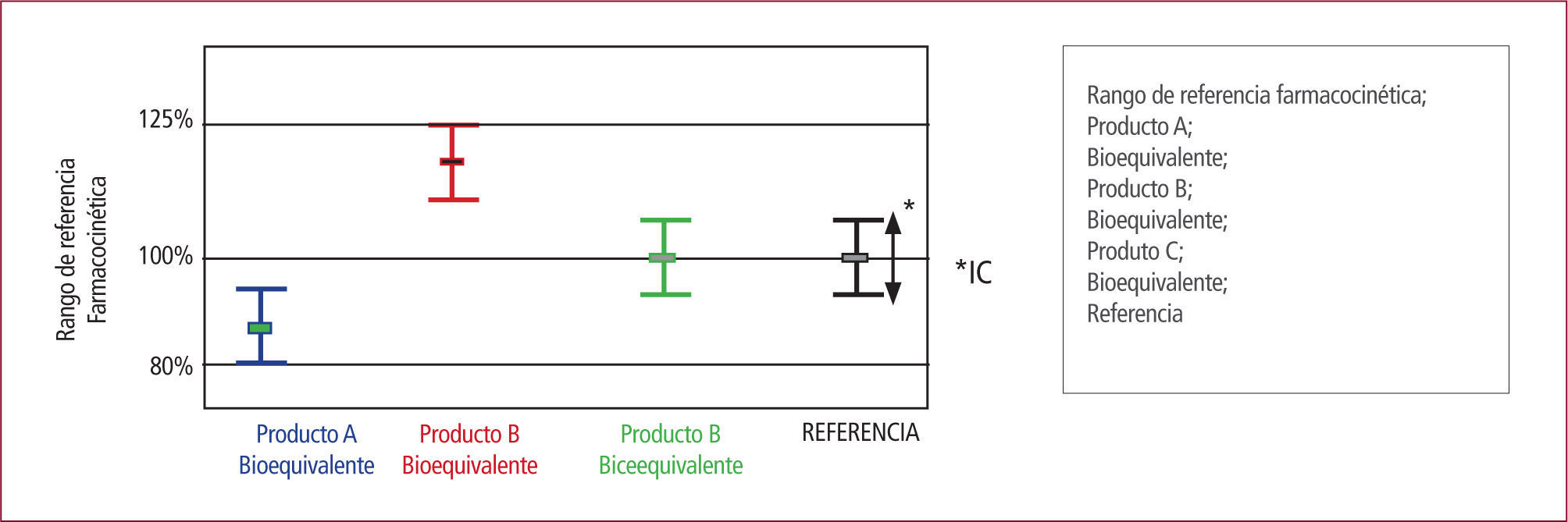
Para las autoridades sanitarias, para ser considerados bioequivalentes, los parámetros farmacocinéticos representados por el área bajo la curva (ABC) y la concentración máxima (Cmáx) del fármaco similar o genérico y su respectivo producto de referencia, deben estar contenidos en el intervalo de confianza del 90% (IC del 90%) dentro del límite del 80% al 125%. Esto significa que para ser aceptados como bioequivalentes, los valores extremos del intervalo de confianza del 90% para la relación entre la media geométrica del ABC prueba/ABC referencia y Cmáx prueba/Cmáx referencia debe ser > 0,8 y <1, 25 (Figura 1).

Comparación de los rangos terapéuticos de tres formulaciones farmacéuticas
Figura 1. El producto A será bioequivalente al de referencia, ya que el Intervalo de Confianza del 90% de su relación ABC prueba/ABC referencia y Cmáx prueba/Cmáx referencia está en el intervalo del 80-125%. El producto B no será bioequivalente, ya que el Intervalo de Confianza del 90% de su ABC prueba/ABC referencia y Cmáx prueba/ Cmáx referencia está fuera de los límites del 80-125%.
La bioequivalencia, en la mayoría de los casos, particularmente para el tratamiento de condiciones agudas, asegura que el medicamento genérico es terapéuticamente equivalente al producto de referencia; es decir, tiene la misma eficacia clínica y seguridad. La bioequivalencia certifica que ambos tienen la misma biodisponibilidad, definida como la cantidad y la velocidad a la que el ingrediente activo es absorbido a partir de una fórmula farmacéutica y se convierte en disponible en el sitio de acción. Los productos que tienen su bioequivalencia y biodisponibilidad demostradas serán considerados intercambiables. La intercambiabilidad es la propiedad que garantiza al paciente calidad, seguridad y eficacia cuando se cambia el producto de referencia por un medicamento bioequivalente a él, considerado terapéuticamente equivalente (1).
En la práctica clínica, una de las cuestiones más importantes y discutidas con el objetivo de minimizar los costes es la sustitución de las fórmulas farmacéuticas. Esta cuestión es particularmente importante en el tratamiento de enfermedades crónicas y graves tales como la epilepsia (2-4). ¿La modificación de las fórmulas puede ser peligrosa para los pacientes con trastornos como la epilepsia? ¿Tal sustitución podría poner en peligro la seguridad y/o eficacia del tratamiento? ¿Es necesaria la monitorización cuando de hecho se cambian las fórmulas?
La respuesta a estas preguntas requiere el análisis de situaciones tales como las que se presentan en la Figura 2.
 por un fármaco de biodisponibilidad alta (B). Esta sustitución puede promover la ocurrencia de eventos adversos, en este caso manifestados por toxicidad. En la situación opuesta, podrían aparecer crisis epilépticas.")
Comparación de los rangos terapéuticos de cuatro formulaciones farmacéuticas
Figura 2. En teoría, es posible que un paciente presente un incremento de casi un 50% en la concentración sérica cuando cambia un fármaco genérico de baja biodisponibilidad (A) por un fármaco de biodisponibilidad alta (B). Esta sustitución puede promover la ocurrencia de eventos adversos, en este caso manifestados por toxicidad. En la situación opuesta, podrían aparecer crisis epilépticas.
En una extensa revisión de la literatura, Dios et al. (5) encontraron que la mayoría de los estudios sobre la sustitución de los fármacos antiepilépticos (FAEs) consiste en informes de casos clínicos o series de casos descritos por la presencia de eventos adversos o descompensación clínica representada por el incremento de las crisis en el momento de la sustitución. En la actualidad, existen pocos estudios controlados y la evidencia disponible se basa principalmente en la opinión de expertos. Sin embargo, hay evidencias de problemas cuando se producen cambios de fórmulas farmacéuticas en la epilepsia. En un estudio con 251 pacientes a los que se les sustituyó sus FAEs por genéricos, Crawford et al. (6) reportaron que el 10,8% de ellos tenía un problema atribuible a este cambio confirmado y el 9,9%, un problema no confirmado. Estos autores sugirieron que los efectos negativos de la prescripción de un genérico se sobreponían al ahorro en los costes (6).
Los FAEs tradicionales tienen características que los hacen difíciles de reemplazar, representadas por índice terapéutico estrecho, poca solubilidad en agua y cinética no lineal.
Para entender el significado de índice terapéutico estrecho es necesario introducir el concepto de índice terapéutico. Índice terapéutico (IT) es la relación de la concentración del fármaco eficaz en el 50% de los sujetos (CE50) y la concentración tóxica para el 50% de estos (CT50). Por lo tanto, IT= CE50/CT50. Algunos fármacos antiepilépticos tienen un rango estrecho en el que se controlan las crisis sin toxicidad. El “rango terapéutico” de un FAE es un intento de trasladar el concepto experimental de IT a la clínica. Su uso en la práctica clínica es limitado y puede inducir a errores en pacientes individuales. Muchos pueden tolerar y requerir concentraciones séricas superiores al rango terapéutico habitual, mientras que otros conseguirán el control de crisis o presentarán eventos adversos en concentraciones de FAEs por debajo de sus límites inferiores.
Un fármaco es considerado como de índice terapéutico estrecho cuando la relación entre la concentración tóxica más baja, en la cual se produce comúnmente toxicidad clínica, y la concentración que proporciona efecto terapéutico es ≤ 2. Ejemplos de tales FAEs son fenitoína [IT = concentración tóxica mínima (20 mg/L)/concentración efectiva (10 mg/L) = 2]; carbamacepina [IT = concentración tóxica mínima (8 mg/L)/concentración efectiva (4 mg/L) = 2] y el ácido valproico [IT = concentración tóxica mínima (100 mg/L)/concentración efectiva (50 mg/L) = 2].
Para un medicamento de IT estrecho y/o con el metabolismo saturable, sería más conveniente exigir límites de bioequivalencia más estrechos. La fenitoína es un FAE con problemas terapéuticos potenciales por alteraciones en su biodisponibilidad cuando cambia sus fórmulas farmacéuticas por su baja solubilidad en agua, por su metabolismo saturable y IT estrecho. Tales problemas se revelaron en 1968, cuando, en Australia, ocurrió un brote de intoxicación por fenitoina en el momento del intercambio del excipiente de calcio sulfato por lactosa en la formulación de referencia. Como resultado, hubo aumento sustancial en la biodisponibilidad y en la concentración sérica de fenitoína hasta 80% a 100%, de modo que 51 pacientes presentaron ataxia, diplopía y vómitos con concentraciones de fenitoína sérica por encima de 20 mg/L. Con el regreso del excipiente original, ocurrió una remisión completa de los síntomas (7).
Las diversas fórmulas de fenitoína difieren con respecto a varios factores: la forma de la sal (sal de sodio o ácido libre), fórmula (comprimido, cápsula, suspensión), contenido de fenitoína, tamaño y forma de sus partículas, y otras características de la formulación (aglutinantes, excipientes, lubricantes, etc.), tiempo de disolución y velocidad de desintegración, entre otros. Todas estas características afectan a la biodisponibilidad de las formulaciones de fenitoína y deben ser consideradas en cada cambio de una fórmula para otra (8-10).
La carbamacepina también es insoluble en agua, tiene IT estrecho y cinética parcialmente no lineal mientras que el valproato parece ser más fácilmente sustituido, ya que es soluble en agua y no satura sus vías metabólicas. Sin embargo, el ácido valproico también tiene IT estrecho. Hay un informe incidental de recurrencia de crisis epilépticas después de reemplazar Depakene® por una formulación genérica de ácido valproico (11). Sin embargo, no hubo diferencias en la biodisponibilidad o en el control de crisis en el cambio de una fórmula de referencia de ácido valproico y una fórmula genérica en 64 pacientes con retraso mental (12). En un reporte de caso, Sherwood et al. (13) informaron un aumento del número de efectos adversos gastrointestinales después de cambiar una fórmula genérica de ácido valproico.
La demostración de la bioequivalencia por los FAEs no significa intercambiabilidad. Un ejemplo es lo que ocurrió con dos formulaciones de fenitoína que habían demostrado ser bioequivalentes en sujetos sanos, en ayunas. Cuando la misma dosis de fármaco (100 mg) se ingirió después de una comida grasa, hubo una diferencia del 13% en la biodisponibilidad relativa. Sin embargo, debido a su cinética no lineal, se calculó que 13% de esta diferencia en la biodisponibilidad causó una reducción del 37% en las concentraciones plasmáticas de los niveles de fenitoína. Consecuentemente, hubo una caída por debajo del “rango terapéutico” en el 46% de los pacientes (14). Del mismo modo Mayer et al. (15) demostraron diferencias significativas con dos fórmulas de carbamacepina administradas a 13 pacientes. Aunque bioequivalentes en términos de farmacocinética, ocho de ellos presentaron eventos adversos como mareos, náuseas, ataxia, diplopía, nistagmo después de cambiar las fórmulas.
Los nuevos FAEs no tienen el “perfil de riesgo” de los medicamentos afectados por problemas de biodisponibilidad como insolubilidad en agua y cinética no lineal. Lamotrigina, oxcarbacepina, gabapentina y topiramato también no tienen IT estrecho. Sin embargo, la relación entre las concentraciones plasmáticas y los efectos clínicos de los FAEs no se han establecido completamente. La cinética de estos últimos es lineal (con excepción de la gabapentina, que satura las enzimas responsables de su absorción) y la absorción oral es satisfactoria. Hay indicios de que pueden ocurrir problemas cuando sus formulaciones son sustituidas (16). Es posible que aquí el problema se produzca debido al concepto de umbral terapéutico estrecho individual. Según este concepto, los individuos con alta propensión a crisis solamente alcanzarán el control de las mismas en los límites superiores de los rangos terapéuticos. Esto significa que estrechan su margen terapéutico de los FAEs, incluso de aquellos con gran índice terapéutico.
Andermann et al. (16) compararon productos de marca con genéricos en un estudio cuyos objetivos fueron (a) cuantificar y comparar las tasas de retorno al producto original de FAEs en las farmacias después de haber sido cambiados por FAEs genéricos u otros fármacos, y (b) evaluar las implicaciones clínicas del cambio de una fórmula de lamotrigina de marca (Lamictal®) a una de lamotrigina genérica y comprobar si aparecen signos sugerentes de sus consecuencias. La lamotrigina fue uno de los primeros FAEs de nueva generación que tuvo versiones genéricas. En este estudio, los autores utilizaron datos de farmacias públicas de la provincia de Ontario en Canadá y calcularon las tasas de retorno de una fórmula genérica de un FAE a una fórmula de marca [Lamictal®, Frisium® (clobazam), y Depakene® (ácido valproico)] comparándolas con otros fármacos utilizados crónicamente como para el tratamiento de hiperlipidemia (Zocor®; sivastatina) y depresión (Prozac® (fluoxetina) y Celexa® (citalopram), desde enero de 2002 hasta marzo de 2006. Investigaron también la demanda de estos medicamentos en la farmacias y las dosis de FAEs en pacientes tratados con lamotrigina que regresaron a la fórmula de marca, es decir, Lamictal®, en comparación con aquellos que continuaron con la formulación genérica. De los 1.354 pacientes a los que se prescribió lamotrigina genérica, el 12,9% volvieron al Lamictal®. Las tasas de retorno de los otros FAEs fueron aproximadamente el 20% para el clobazam y el ácido valproico. Las tasas de retorno entre los FAEs fueron sustancialmente más altas que las de otros medicamentos no antiepilépticos (de 1,5 a 2,9%) (Figura 3).
: 464-469, 2007.")
Comparación de fármacos antiepilépticos, antidepresivos y antilipidémico
Figura 3. Cambio de los medicamentos para volver a la fórmula de marca después de la administración de uno o más genéricos en la provincia de Ontario, Canadá. Modificado de Andermann et al. Epilepsia 48(3): 464-469, 2007.
Se observó además un aumento significativo de las dosis de lamotrigina después de la sustitución del producto de marca por genéricos en los que no regresaron al medicamento de marca (6,2%, p <0,0001). Los resultados de este estudio interesante reflejan la dificultad para aceptar el intercambio de FAEs de marca por los compuestos genéricos. Estos resultados también pueden ser indicativos de lo que puede haber sido un aumento de la toxicidad y/o pérdida de control de las crisis epilépticas después del cambio a fórmulas genéricas de los FAEs.
También con la lamotrigina se llevó a cabo en Dinamarca un estudio farmacocinético que incluyó seis pacientes en los que fue posible comparar los patrones farmacocinéticos de las fórmulas de referencia de la lamotrigina y diversas formulaciones genéricas (17). Eran pacientes que habían informado problemas con el cambio de formulaciones o cuyos médicos sospecharon de su ocurrencia por constatar signos de intoxicación o falta de eficacia, es decir, la aparición de crisis por ocasión del cambio. En estos seis pacientes fueron verificados perfiles de los niveles séricos diarios de lamotrigina en muestras de suero recogidas cada 3 o 4 horas durante 24 horas, lo que permitió la evaluación de los parámetros: Cmáx, Cmin, Cmáx/min y Cx. Uno de los casos estudiados fue el de un paciente de 25 años con deterioro neurológico y epilepsia con crisis focales y generalizadas que usaba lamotrigina en monoterapia, en la dosis de 500 mg/día. Se quejó de que, después de cambiar la fórmula de referencia por una formulación genérica, presentó mayor inestabilidad para caminar y se cayó, presentando fractura del cráneo y hematoma epidural. La sospecha clínica fue de ataxia debido al aumento de la biodisponibilidad de la preparación. Los estudios farmacocinéticos de la formulación de referencia y de la fórmula genérica anteriormente administrada después de dos semanas de uso mostraron que la biodisponibilidad de la formulación genérica fue significativamente más alta. El diagnóstico clínico final fue de ataxia causada por la formulación genérica (17) (Figura 4).

Es importante destacar que debido a la demostración de las graves consecuencias de la sustitución de los antiepilépticos, las autoridades sanitarias del gobierno danés estrecharon los límites para bioequivalencia de los FAEs para 90 hasta 111% y eliminaron la obligación de los farmacéuticos de considerar intercambiables los FAEs de los pacientes con índice terapéutico estrecho el cual, para la lamotrigina, fue establecido como niveles plasmáticos por encima de 30 mg/ml (18).
La American Academy of Neurology (19) recomienda que los medicamentos no deben ser sustituidos a menos que sea “médicamente necesario” y aconseja estricto control de los niveles plasmáticos y de los resultados clínicos con ocasión del reemplazo, mientras se lleva a cabo la sustitución. La Epilepsy Foundation of America (20) sostiene que el individuo y su médico deben dar su consentimiento antes del reemplazo de la medicación, sea la de un producto de referencia por un genérico o de un medicamento genérico por otro genérico. La Food and Drug Administration incentiva a los médicos estadounidenses y a las personas con epilepsia a reportar las crisis epilépticas que se producen con la sustitución de sus medicamentos.
Como se ha explicado, la sustitución de los fármacos de referencia por los genéricos o viceversa puede causar problemas como toxicidad o reducción de los niveles séricos y crisis. Ambos pueden aumentar significativamente los costos y las consecuencias de esta actitud terapéutica (21). Por lo tanto, aunque los costos de los medicamentos genéricos o similares sean inferiores a los de productos de referencia porque a los fabricantes se les eximen los relacionados con los estudios de investigación y los registros iniciales, los estudios de análisis de costo de reemplazo, por ejemplo de la carbamacepina de referencia por la carbamacepina genérica, rechazan estas ventajas. Argumosa y Herranz (22) demostraron que el ahorro inicial en el sistema de salud de España excedió mucho por el lado de los costos, ya sea por ineficacia (crisis recurrentes) o por toxicidad (efectos adversos). La ocurrencia de una sola crisis en una persona con crisis controladas puede causar efectos devastadores, como accidentes de tráfico, pérdida de empleo, lesiones e incluso la muerte. El médico debe estar siempre atento a los cambios en la prescripción y los farmacéuticos siempre alerta cuando estos, de hecho, ocurran. Más importante aún, es que el paciente sea educado sobre los principios que rigen los aspectos de producción, comercialización y de la farmacocinética de las diferentes fórmulas de los FAEs.
Finalmente, los pacientes con epilepsia pueden recibir una formulación genérica para el tratamiento de sus crisis. Siempre hay que recordar que los medicamentos genéricos son bioequivalentes al medicamento de referencia, lo que no significa que los medicamentos genéricos sean bioequivalentes entre sí. Por otra parte, este proceso abre la posibilidad de que la medicación sea cambiada repetidamente por formulaciones similares o de otros genéricos porque las farmacias pueden recibir productos de diversos laboratorios. Por último, durante el ingreso del paciente en un hospital, puede incluso ser utilizada otra formulación diferente a las anteriores. Por estas razones, se debe reconocer que al iniciar el tratamiento con una formulación similar o genérica muy probablemente se iniciará la sustitución de fármacos de uno o más fabricantes en algún momento futuro, lo que producirá ineficacia y crisis o eventos adversos, como señales de intoxicación (21).
La autora declara no tener conflictos de interés, con relación a este artículo.