



El ojo, además de entregarnos el sentido de la visión, también nos permite conocer la salud general del organismo. Muchas enfermedades sistémicas se manifiestan en el órgano visual antes, durante o después del debut de ellas a nivel sistémico. Este compromiso visual es variado y depende de la enfermedad en cuestión, destacando la escleritis, uveítis y vasculitis retinales. Conocer el estado ocular permitirá al clínico realizar diagnósticos más asertivos y oportunos, realizar el tratamiento más adecuado y definir pronóstico en varias enfermedades, que tienen al globo ocular como un órgano blanco.
El objetivo de esta revisión es atraer la atención del lector sobre el compromiso oftalmológico en varias enfermedades.
The eye besides giving us sight, it let us know the general health of the whole body as well. Many systemic diseases become evident in the visual organ before, during or after its systemic onset. The visual involvement in these diseases is wide and depends on the underlying disease. It is worth to stand out the escleritis, uveitis and retinal vasculitis. The knowledge of the ocular health will let the physician to diagnose more accurately and in time, to start the most appropriate treatment and to define the prognosis of several diseases that have the eye as a target organ.
The aim of this review is to draw the reader's attention to the ophthalmic involvement in several systemic diseases.
La importancia de la visión y su relación con el bienestar general es conocida desde la Antigüedad. Cicero (106 – 73 AC) señaló: “Ut imago est animi voltus sic indices oculi” (La cara es un cuadro de la mente; los ojos, su intérprete). Más adelante, a mediados del siglo XIV, se habría originado este conocido proverbio: “los ojos son la ventana del alma”.
El examen de los ojos, no sólo nos muestra la expresión de sentimientos y estados de ánimo del ser humano, sino que también nos permite conocer padecimientos que afectan a todo su organismo.
El conocimiento del compromiso ocular en varias enfermedades sistémicas ayudará al clínico tanto en los procesos diagnósticos, terapéuticos y pronósticos.
En este artículo nos referiremos a algunas enfermedades sistémicos que dan manifestaciones en el tracto uveal, excluyendo las de origen infeccioso: bacterianas como tuberculosis, lepra y sífilis; micóticas como candidiasis, criptocococis e histoplasmosis; parasitarias como cisticercosis, toxocariasis, toxoplasmosis y oncocercosis; y virales tales como herpes simplex, zoster, y citomegalovirus. Síndromes enmascarados como leucemias, linfomas, melanomas, retinoblastomas, síndromes metastásicos y paraneoplásicos merecen un capítulo aparte y no serán tratados en esta revisión.
El compromiso inflamatorio ocular puede ocurrir en diversos segmentos del globo. La orientación diagnóstica se basa en la localización principal de la afectación uveal. Basado en este concepto, se ha llegado al consenso de clasificar a las uveitis es: Anteriores, Intermedias, Posteriores y Panuveitia (Tabla 1).
Clasificación Anatómica De Las Uveitis
| Tipo | Sitio Primario de Inflamación | Incluye |
|---|---|---|
| Uveítis Anterior | Cámara Anterior | Iritis Iridociclitis Ciclitis anterior |
| Uveítis Intermedia | Vitreo | Pars planitis Ciclitis posterior Hialitis |
| Uveítis Posterior | Retina o Coroides | Coroiditis focal, multifocal o difusa Corioretinitis Retinocoroiditis Retinitis Neuroretinitis |
| Panuveitis | Cámara Anterior, Vitreo y Retina o Coroides |
Jabs DA, Nussenblatt RB, Rosembaum JT. Standarization of Uveitis nomenclature (SUN) Working Group. Standarization of uveitis nomenclature for reporting clinical data. Results from the First International Workshop. Am J Ophthalmol 2005; 140:509–516
A continuación se describen algunas enfermedades en las cuales el examen del tracto uveal, puede dar claves relevantes en torno al diagnóstico, tratamiento y pronóstico de éstas (Tabla 2).
Compromiso Oftalmológico De Enfermedades Sistémicas
| Enfermedad | Frecuencia de manifestaciones oftalmológicas | Principales manifestaciones oftalmológicas | |
|---|---|---|---|
| Behçet | 90% en hombres 70% en mujeres | Uveítis anterior Vasculitis retinal, Viteritis Neovascularización retinal y coroídea Papilitis | |
| HLA B27 + | Reiter | 10% | Conjuntivitis Uveitis anterior Epiescleritis Escleritis |
| EAA | 25% | ||
| EEI | CU: 5% EC: 3% | ||
| Artropatía psoriática | 20% | ||
| Enfermedades del tejido conectivo | AR | <1% del tipo inflamatorio 15–20% ojo seco | Ojo seco (Asociado o no a Sd. de Sjögren) Queratitis ulcerativa Epiescleritis, escleritis Uveitis anterior Vasculitis (en LES, GW y PAN) Neuropatía óptica |
| LES | 30% ojo seco Raro tipo inflamatorio | ||
| AJI | Pauciarticular: 20% Poliarticular: 5–10% Sistémica: <5% | ||
| Vasculitis sistémicas | GW: 50% | ||
| PAN: 20% | |||
| Sarcoidosis | 25–50% | Ojo seco Uveitis anterior, intermedia, posterior y difusa Vasculitis, papilitis Granulomas retinales y coroídeos | |
| Síndrome de Vogt Koyanagi Harada | 100% | Uveitis difusa, Poliosis Desprendimiento seroso de retina, Papilitis | |
EEA: Espondiloatritis anquilosante; EEI: Enfermedad inflamatoria intestinal; CU: Colitis ulcerosa; EC: Enfermedad de Crohn; AR: Artritis reumatoidea; LES: Lupus eritematoso sistémico; AJI: Artritis juvenil idiopática; GW: Granulomatosis de Wegener; PAN: Poliarteritis nodosa.
Especialidades comprometidas: Dermatología, neurología, psiquiatría, reumatología, gastroenterología, cardiología, urología, cirugía vascular, odontología, oftalmología.
La Enfermedad de Behcet es una vasculitis necrotizante no granulomatosa, multisistémica, recurrente, de etiología desconocida. Observándose con mayor frecuencia compromiso oftalmológico, dermatológico, reumatológico y neuropsiquiátrico.
Fue Hipócrates quien posiblemente describió por primera vez una asociación entre inflamación ocular y lesiones orales y genitales. No obstante, esta enfermedad lleva el nombre de Hulusi Behcet (1889–1948), dermatólogo turco, quien en 1937 describió esta enfermedad, reportando su triada diagnóstica de inflamación intraocular recurrente con úlceras orales y genitales.
Esta enfermedad tiene una distribución mundial, siendo especialmente común en los países del Lejano Oriente y del Meditárraneo (Japón, Turquía, Italia, Grecia, Israel, Líbano y otros).
En Japón, la EB constituye el 22% de las causas de Uveítis, en Brasil el 2% y en Inglaterra el 3%.
La edad media de comienzo de la enfermedad es entre los 20 a 40 años, pero se han reportado casos en niños y ancianos.
La etiología de esta vasculitis sistémica oclusiva es desconocida, existiendo histopatológicamente una perivasculitis asociada a destrucción celular. La hipótesis más aceptada en relación al daño tisular, ha sido la de una enfermedad mediada por complejos inmunes circulantes, con infiltración predominante de neutrófilos. Últimamente, se ha destacado la importancia de los linfocitos T, encontrándose alteraciones de estos en pacientes con enfermedad ocular. Se han detectado células T inmaduras circulantes, con aumento de la fracción T8 y una disminución de la fracción T4/T8. Los niveles séricos de gamma-interferón están aumentados. El antígeno de histocompatibilidad HLA B51 está asociado con esta enfermedad en pacientes japoneses con un riesgo relativo de 9.4. La frecuencia de esta antígeno en sujetos control fue de 12% y en enfermos con Behcet de 57%. Estudios más recientes utilizando Reacción de cadena polimerasa, encontró factor de riesgo relativo de 7.9 (3).
Dado que la etiología es desconocida y no existiendo exámenes de laboratorio específicos, el diagnóstico debe hacerse en base a los hallazgos clínicos, basándose en los denominados criterios diagnósticos enunciados por el Comité de Investigación de la Enfermedad de Behcet de Japón (4).
Se denominan criterios mayores por su mayor frecuencia de aparición a la triada clásica: úlceras aftosas recurrentes de la mucosa oral, úlceras genitales, enfermedad inflamatoria ocular recurrente anterior y posterior; a lo que se agregó lesiones cutáneas como eritema nodoso, acné, hipersensibilidad cutánea y tromboflebitis.
Se clasifican como criterios menores, no por ser menos severos, sino porque su frecuencia de ocurrencia es significativamente menor a la artritis, úlcera intestinal, epididimitis, enfermedad vascular como tromboflebitis superficial y profunda, obstrucción vascular, aneurismas, aortitis y síntomas neuropsiquiátricos.
Se habla de EB completo cuando están presentes los 4 criterios mayores simultáneamente o durante la evolución, es de tipo incompleto cuando existen 3 criterios mayores simultáneamente o durante la evolución o enfermedad ocular recurrente típica con otro criterio mayor. Se considera sospecha de EB cuando se observan 2 criterios mayores excluyendo el compromiso ocular y sospecha cuando aparece un solo criterio.
Se reconocen tres formas especiales de EB: Behcet intestinal, que puede producir úlceras a cualquier nivel (similar a lo producido en la enfermedad de Crohn); Vásculo Behcet, que incluye tromboflebitis superficial y profunda, trombosis venosas, aneurismas y aortitis, que pueden determinar la muerte del paciente; y Neuro Behcet, cuando la enfermedad involucra al sistema nervioso central, pudiendo aparecer accidentes cerebrovasculares, déficit focales y síntomas neuropsiquiátricos, los cuales le otorgan un matiz de gravedad.
El compromiso oftalmológico es cercano al 80%, siendo más frecuente en el hombre 86,2% que en la mujer (67,8%) (1, 2). Generalmente ocurre 2 ó 3 años luego del inicio de la enfermedad, aunque los síntomas oculares pueden ser la primera manifestación de la enfermedad en el 20% de los casos.
Generalmente ambos ojos están afectados, con cuadro uveal de comienzo agudo, no granulomatoso, que se hace recurrente, con manifestaciones tanto en el segmento anterior, como en el posterior.
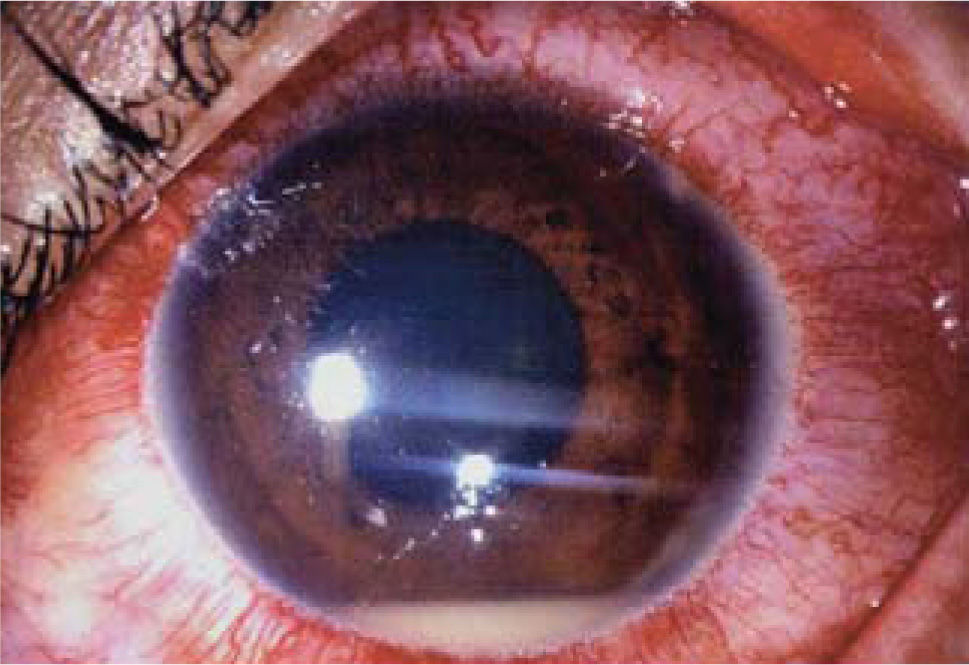
El compromiso del segmento anterior está dado por la presencia de una uveítis aguda intensa que en un tercio de los casos se acompaña de hipopion (pus en la cámara anterior), esta inflamación severa puede llevar a la formación de sinequias, glaucoma y catarata, pero con un ojo relativamente blanco (ver Figura 1).

La afectación del segmento posterior está dado por la enfermedad retinal, la que puede llevar con frecuencia a la ceguera. Los episodios vaso-oclusivos retinales recurrentes se caracterizan por la presencia de vasculitis tanto arterial como venosa, infiltrados y hemorragias retinales, edema de retina, vitreitis, isquemia retinal, neovascularización coroidea y compromiso del nervio óptico.
El diagnóstico es clínico, basado en los criterios previamente descritos. Además, una tipificación positiva para el antígeno de histocompatibilidad humana HLA B51 también orienta al diagnóstico (3).
El tratamiento debe ser instaurado lo más pronto posible, teniendo como objetivo reducir la intensidad de la inflamación y la frecuencia de las crisis, para así prevenir el deterioro visual, que de no ser tratado adecuadamente se hace irreversible, llevando a la ceguera.
El uso de inmunosupresores constituye indicación absoluta como terapia de primera línea.
La uveítis anterior se trata con midriáticos, ciclopléjicos y corticoides tópicos. El compromiso del segmento posterior, es manejado con en el uso de corticoides sistémicos en altas dosis, agentes citotóxicos (alquilantes), colchicina, ciclosporina y últimamente el uso de agentes biológicos. Estos últimos han demostrado efectividad y seguridad en pacientes resistentes a los inmunosupresores habituales.
2Uveitis HLA-B27Especialidades comprometidas: Reumatología, urología, gastroenterología, traumatología, dermatología y oftalmología.
Las siguientes enfermedades tienen como factor en común, la predominancia del fenotipo HLA B27 positivo y el compromiso oftalmológico manifestado como una uveitis anterior (o iritis o iridociclitis). La uveitis anterior se presenta clásicamente con ojo rojo profundo1 doloroso, fotofobia, precipitados queráticos en el endotelio corneal, y células y proteínas flotando en el humor acuoso.
Síndrome de ReiterLa tríada clásica de uretritis inespecífica, artritis y conjuntivitis puede aparecer luego de infección disentérica por gram negativos. 85% de estos pacientes tienen HLA B27 + (5, 6), versus el 7% de la población sana.
El compromiso oftalmológico más frecuente es la conjuntivitis que es generalmente bilateral del tipo papilar con descarga mucopurulenta (30–60%) (3). Algunos pacientes pueden desarrollar queratitis que en raros casos puede dejar cicatrices corneales. Posee cultivos negativos y normalmente cede espontáneamente dentro de 1 a 2 semanas.
La afectación uveal generalmente no sobrepasa el 10% (3, 5, 6), no obstante, puede ser un poco mayor en portadores de HLA B27. Consiste en una inflamación del iris y el cuerpo ciliar de tipo aguda no granulmatosa.
El diagnóstico de esta enfermedad se basa en los hallazgos clínicos, apoyado en la presencia del fenotipo HLA B27 positivo.
Mientras que el tratamiento oftalmológico se realiza con esteroides tópicos y agentes midriáticos, el compromiso de otros órganos puede requerir el uso de AINES, esteroides y/o inmunosupresores.
Espondilitis anquilosanteEsta enfermedad se caracteriza por una inflamación crónica de articulaciones del esqueleto axial, principalmente de la sacroilíaca, que lleva a la calcificación y osificación de los ligamentos y cápsulas articulares y finalmente a la anquilosis.
Aproximadamente el 90% de estos pacientes tiene fenotipo HLA B27 positivo (3, 5, 7, 8) y el compromiso oftalmológico ocurre en el 25% de ellos (5, 6, 9).
La uveitis anterior es la principal manifestación ocular de esta enfermedad. Se presenta de forma unilateral recurrente (con compromiso de ambos ojos asincrónicamente), con gran inflamación del segmento anterior que puede llevar a la formación de hipopion. Otras manifestaciones menos frecuentes son queratitis numular, escleritis1, epiescleritis2, papilitis y edema de retina. Si no es tratada oportunamente pueden aparecer sinequias posteriores, catarata y glaucoma, entre otros.
El tratamiento oftalmológico se basa en el control de la enfermedad de base que se puede realizar tanto con antiinflamatorios esteroidales y no esteroidales como inmunosupresores. La terapia local incluye el uso de esteroides tópicos y perioculares, con agentes midríaticos.
Enfermedad inflamatoria intestinalEste grupo incluye tanto a la colitis ulcerosa (CU) como la enfermedad de Crohn (EC). Corresponden a enfermedades inflamatorias crónicas recurrentes del tubo digestivo, de etiología no conocida completamente. Si bien se han descrito algunos genes asociados a estas entidades, estudios observacionales sugieren que podrían ser gatillados por fenómenos ambientales como gérmenes o estrés (10). Patogénicamente ambas presentan activación de linfocitos TCD4 de la mucosa del tubo digestivo, sugiriendo una pérdida de la tolerancia a antígenos externos provenientes de alimentos o de la flora comensal (tolerancia oral).
El compromiso oftalmológico es de aproximadamente 5% para la CU y de 3% para la EC (3, 6). La manifestación más frecuente es una uveítis aguda no granulomatosa anterior, aunque también se han descrito infiltrados corneales, epiescleritis y escleritis (11). En raras ocasiones puede existir afectación del segmento posterior en forma de papilitis, coroiditis multifocal y vasculitis retinal.
El tratamiento para el compromiso ocular se basa en el uso de esteroides tópicos, no obstante puede requerirse terapia sistémica dependiendo de estado inflamatorio intestinal que presente el paciente.
Artropatía psoriáticaLa uveítis anterior es rara en pacientes con psoriasis. No obstante, el 20% de pacientes con psoriasis puede desarrollar uveitis, enfermedad inflamatoria intestinal o sacroileitis, cuando se asocia a artritis.
El uso de esteroides tópicos y midriáticos son la base del tratamiento del compromiso del sistema visual.
3Enfermedades del tejido conectivoArtritis reumatoidea (AR)Enfermedad inflamatoria crónica multisistémica caracterizada por una sinovitis distal simétrica, destructiva y deformante. El compromiso extra articular ocurre en pacientes con títulos altos de factor reumatoídeo y anticuerpos anti péptido cíclico citrulinado. Consistente en nódulos reumatoídeos, vasculitis reumatoidea, manifestaciones pleuropulmonares, neuropatías, osteoporosis y pancitopenia.
Si bien su etiología es desconocida, se asocia fuertemente a la presencia de HLA DR1, DR3, DR4, DR9 (más frecuente en Chile) y DR10.
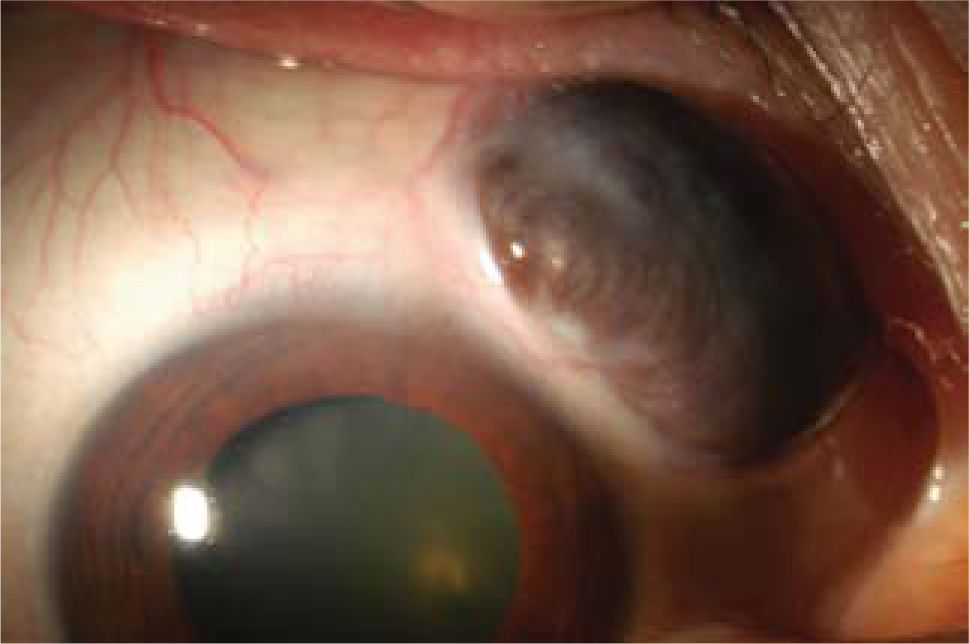
El compromiso oftalmológico ocurre en menos de 1% de los pacientes con AR, principalmente en aquellos con enfermedad de larga data y con presencia de nódulos. Este se caracteriza por queratitis ulcerativa, epiescleritis y escleritis (ver Figura 2). Además, entre un 15 y 20% de los pacientes con AR desarrolla un Síndrome de Sjögren, con los síntomas de ojo seco asociados (10).

El tratamiento se centra en el control de la enfermedad de base por el reumatólogo. Para el compromiso ocular se usan tanto antiinflamatorios esteroidales como no esteroidales en colirio. Ante un proceso inflamatorio más severo, están indicados antiinflamatorios y esteroides sistémicos, así como inmunosupresores. Especial atención merecen los agentes que bloquean la actividad del Factor de Necrosis Tumoral o Anti-TNF. La literatura arroja cada vez más resultados que apoyan el uso de este tipo de fármacos en pacientes con artritis reumatoidea.
El manejo del ojo seco, se basa en el uso de lubricantes oculares y tapones de puntos lagrimales, y uso de ciclosporina tópica para los casos más complejos.
Lupus eritematoso sistémico (LES)Corresponde a una enfermedad autoinmune sistémica en la que el daño tisular es producido por autoanticuerpos y complejos inmune circulantes, así como fenómenos trombóticos y vasculíticos.
Dentro de las manifestaciones sistémicas se encuentran: síntomas cutáneos como rash facial en mariposa, rash discoide, vasculitis, telangectasias, fenómeno de Raynaud y alopecía; artritis, miositis y tendinitis; pericarditis, miocarditis y endocarditis; glomérulonefritis; pleuresía y atelectasias; pancitopenia; esplenomegalia y linfadenopatías; polineuritis, paresias de nervios craneales, epilepsia y psicosis.
El 30% de pacientes con LES presentará síntomas relacionados con ojo seco (pudiendo formar parte o no de un sindrome de Sjögren secundario) (12).
A nivel de los párpados puede existir un rash similar al del lupus discoide. En muy raras ocasiones se puede manifestar con masas orbitarias y edema periorbitario.
En el polo anterior, además de la queratopatía sicca (ojo seco) pueden existir erosiones corneales recurrentes y más raramente queratitis ulcerativa periférica, la que es un marcador ominoso de vasculitis sistémica. La escleritis y epiescleritis son también formas características de manifestación del LES en el ojo. La escleritis ya sea nodular o difusa, traduce actividad vasculítica.
La uveítis anterior rara vez ocurre asilada, siendo más frecuentemente vista en asociación con escleritis.
La afección del segmento posterior del ojo es la que presenta más riesgo de complicaciones que amenacen permanentemente la visión.
La retinopatía lúpica se caracteriza por la presencia de manchas algodonosas (isquemia de la capa de fibras retinales), hemorragias, tortuosidad venosa y adelgazamiento arteriolar, muy similar a las retinopatías diabética e hipertensiva. Por otro lado, los fenómenos trombóticos pueden producir obstrucciones arteriales y venosas retinales, más frecuentes en pacientes con títulos elevados de anticuerpos antifosfolípidos. Las alteraciones vasculares previamente señaladas pueden terminar en una retinopatía isquémica proliferativa, con la neoformación de neovasos, hemorragia vítrea y desprendimiento de retina.
La coroides puede también presentar fenómenos isquémicos evidenciables solamente con estudios angiográficos.
El compromiso del nervio óptico incluye a la neuritis óptica y a la neuropatía óptica isquémica, ambas con riesgo inminente de pérdida permanente de visión. Pueden ser producidos tanto por la obstrucción de vasos arteriolares, vasculitis, fenómenos trombóticos focales asociados a la presencia de anticuerpos antifosfolípidos, y la acción de anticuerpos antineuronales.
El tratamiento del ojo seco es similar a lo descrito previamente. La epiescleritis normalmente es autolimitada, aunque se puede usar esteroides tópicos. Las escleritis por su parte son tratadas con esteroides tópicos en conjunto con AINES sistémicos, reservando el tratamiento esteroidal sistémico e inmunosupresor para los casos más severos o relacionados con vasculitis. El manejo de la uveítis anterior no difiere de lo señalado en las patologías antes descritas.
El compromiso del polo posterior sugiere una mayor gravedad del cuadro y el tratamiento consiste en la administración de corticoides sistémicos con o sin inmunosupresores. Cuando el compromiso vascular de la retina se complica con una retinopatía isquémica se indica fotocoagulación láser.
Artritis juvenil idiopáticaEsta entidad se caracteriza por la inflamación articular de al menos 6 semanas de duración en niños menores de 16 años. El compromiso oftalmológico, depende de la forma de presentación, y es independiente al grado de afectación articular (5, 6, 13).
Según su forma de presentación se clasifica en tres grupos:
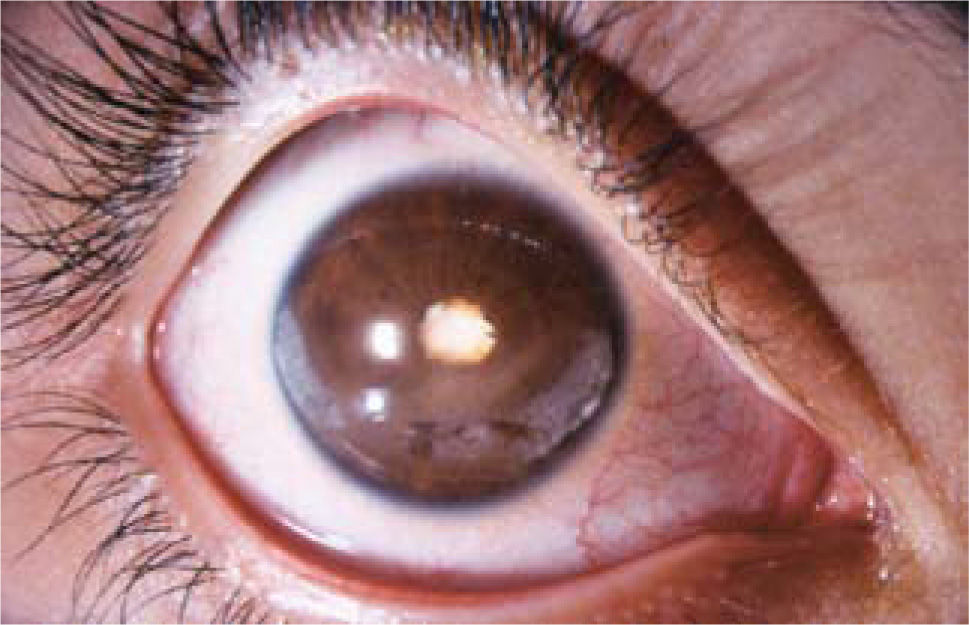
aComienzo pauciarticular (20% de los pacientes presenta uveitis anterior)Es la forma más habitual. Las articulaciones afectadas son máximo 4, siendo la rodilla la articulación más frecuentemente comprometida. El 75% de estos niños son ANA +. Se pueden distinguir 2 subgrupos. El tipo 1, que afecta principalmente niñas menores de 5 años típicamente ANA+ y se manifiesta con uveitis anterior crónica muy poco síntomatica. Por este motivo acuden tardíamente al oftalmólogo con complicaciones como queratopatía en banda (depósito de calcio en la cornea), catarata y glaucoma ya establecidos (ver Figura 3). El tipo 2 por su parte afecta a niños mayores con AAN – y se presenta con cuadros agudos recurrentes y sintomáticos. Este grupo puede presentar hasta un 75% de fenotipo HLA B27+.
bComienzo poliarticular (5–10% cursa con uveítis anterior), con sinequias posteriores y catarata en paciente con AJI.")
Debuta con artritis de 5 ó más articulaciones. Ocurre principalmente en niñas de cualquier edad con un AAN positivo en el 40%.
cComienzo sistémico (Menos del 5% presentará uveitis)Se caracteriza por un compromiso articular mínimo o ausente, pero con síntomas sistémicos como fiebre, rash, linfadenopatías y hépatoesplenomegalia. No hay predominio de género y ocurre a cualquier edad durante la niñez.
Dado que el compromiso inflamatorio oftalmológico es frecuente, la Academia Americana de Pediatría sugiere realizar controles cada 3–4 meses a niños menores de 7 años con comienzo pauci o poliarticular y AAN +; cada 6 meses a niños de cualquier edad con comienzo pauci o poliarticular y AAN -; y cada 12 meses en pacientes con comienzo sistémico.
El tratamiento del compromiso oftalmológico va relacionado con el control de la enfermedad sistémica. El tratamiento esteroidal local es la primera línea de tratamiento para el compromiso ocular. Por otro lado, el uso de esteroides sistémicos, si bien es efectivo, es evitado al máximo en pacientes de este grupo etáreo. Por este motivo, tenemos un bajo umbral para iniciar terapia como metotrexato o micofenolato mofetil como fármacos de segunda línea.
Nuevamente los anti-TNF tienen un rol destacado como terapia alternativa en niños con AJI. Si bien el etanercept es útil en pacientes con AJI, desde el punto de vista oftalmológico la experiencia nos muestra que el Infliximab y Adalimumab son más efectivos en el control de la uveitis.
4Vasculitis sistémicas: Granulomatosis de Wegener (GW) y Poliarteritis nodosa (PAN)Ambas entidades se caracterizan por una vasculitis sistémica necrotizante con compromiso multiorgánico.
La afección ocular ocurre aproximadamente en un 50% de los casos para la GW y en un 20% para la PAN. Estos pacientes pueden presentar escleritis necrotizante, queratitis ulcerativa, uveítis anterior e inflamación orbitaria. La vasculitis retinal es un hallazgo frecuente, caracterizado por envainamiento arteriolar, hemorragias retinales y manchas algodonosas (14–16). En estos casos es mandatario la realización de una angiografía retinal con el fin de evaluar el estado de la perfusión tisular.
El manejo local se centra en el uso de corticoides tópicos, asociado al tratamiento de la enfermedad de base con inmunosupresores. En caso de isquemia retinal severa, se indica panfotocoagulación con láser.
El rituximab es un anticuerpo monoclonal quimérico que causa la lisis de los linfocitos B (CD20+). Ha sido efectivo cuando la terapia inmunosupresora convencional no es tolerada o no ha sido efectiva en controlar la inflamación.
5SarcoidosisCorresponde a una enfermedad inflamatoria granulomatosa sistémica que puede afectar a casi cualquier órgano, pero son los pulmones, los órganos más frecuentemente comprometidos, seguido por ganglios, piel, ojos e hígado.
El compromiso ocular es bastante amplio y ocurre entre 25 y 50% de los casos (17). A nivel de los párpados y conjuntiva pueden aparecer granulomas. El compromiso de la glándula lagrimal puede llevar a un síndrome de Sjögren secundario. La uveitis anterior granulomatosa con presencia de precipitados queráticos gruesos y nódulos iridianos es la forma de presentación ocular más frecuente.
Una uveítis intermedia, que corresponde a la inflamación de la pars plana y hialoides anterior, puede también ser una manifestación de esta enfermedad.
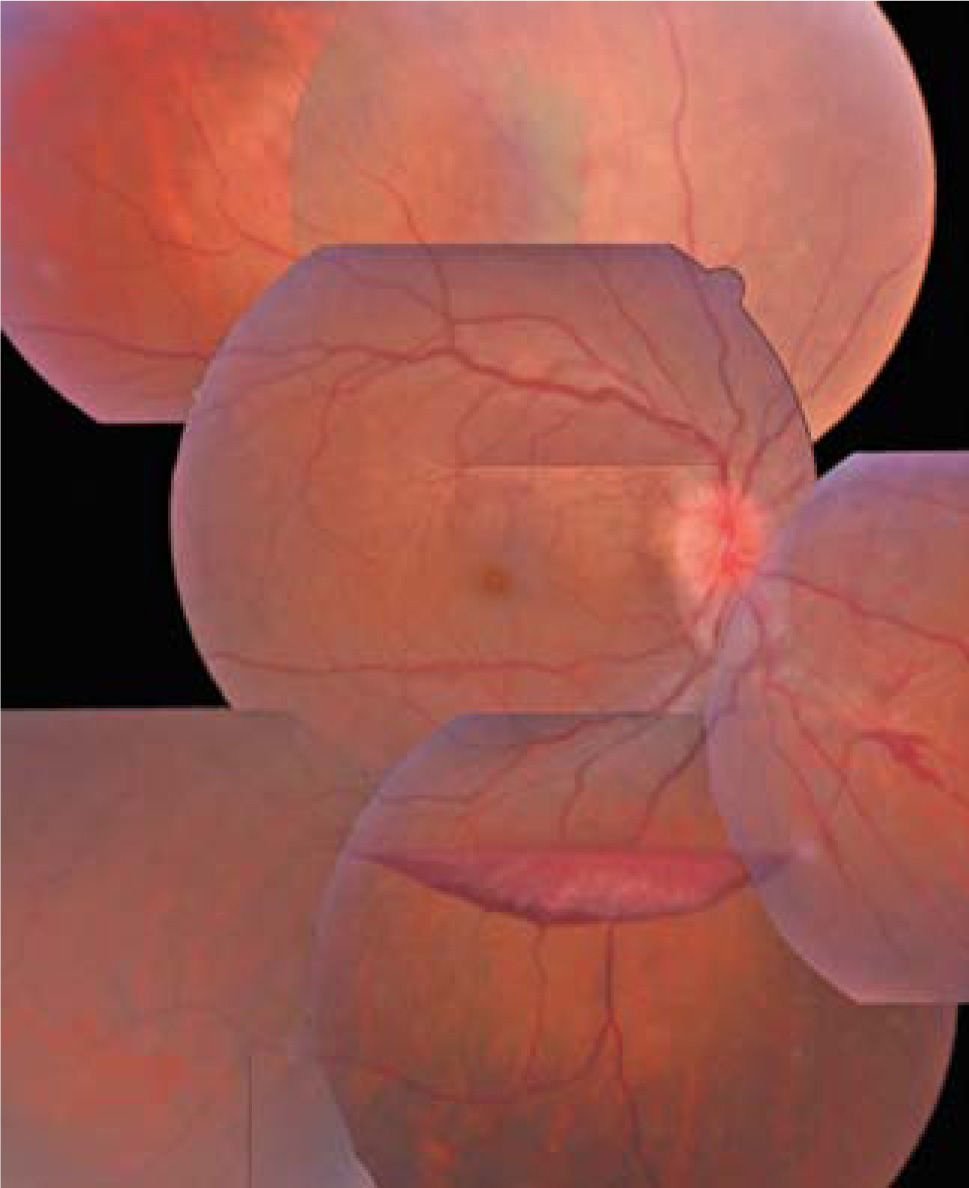
El segmento posterior se compromete con una frecuencia menor (6–33% de pacientes con sarcoidosis), pero con un mayor riesgo de compromiso visual que la afectación del segmento anterior. Ocurre con vasculitis y envainamiento venoso, granulomas generalmente pequeños ubicados en retina y coroides, neovascularización de retina, edema macular y de nervio óptico (ver Figura 4).
.")
Fotomontaje del fondo de ojos de una paciente de 58 años portadora de sarcoidosis. Nótese la hemorragia subhialoidea en forma de bote producida por la rotura de vasos anormales de neoformación secundario a la isquemia retinal producida por la vasculitis. Además en nervio óptico aparece con hiperemia y bordes algo difuminados (papilitis).
Los corticoides tópicos y perioculares, junto con agentes midriáticos son efectivos en el tratamiento de la uveitis anterior, no obstante el compromiso del segmento posterior, puede además requerir el uso de corticoides e inmunosupresores por vía sistémica.
6Síndrome de Vogt Koyanagi Harada (VKH)Especialidades comprometidas: Neurología, reumatología, otorrinolaringología, dermatología, oftalmología.
Es una enfermedad sistémica que compromete diferentes órganos, incluyendo ojos, oídos, piel y meninges. La manifestación clínica más importante de la enfermedad es una uveítis difusa, bilateral, granulomatosa con desprendimiento de retina exudativo y papilitis, acompañada con manifestaciones meníngeas, auditivas y dermatológicas.
Esta enfermedad fue descrita en 1906 por Alfred Vogt. En 1929, Y. Koyanagi describe seis pacientes con iridiciclitis crónica bilateral asociada a poliosis, vitiligo y disacusia. Posteriormente en 1926, Harada caracterizó a una uveítis posterior con desprendimiento de retina exudativo asociado a pleocitosis en líquido cefaloraquídeo. Pero fue J. Babel en 1932 quien sugirió que estos cuadros clínicos correspondían a una misma enfermedad, con diferentes formas de expresión, dependiendo de la intensidad y etapa clínica de su diagnóstico, entidad que denominó Síndrome de Vogt Koyanagi Harada, término como se conoce actualmente.
A pesar de que su distribución es mundial, su incidencia varía de acuerdo al país y en ciertos grupos étnicos y raciales, siendo más frecuentes en razas pigmentadas especialmente en orientales y en América en el indio americano. En Japón representa el 8%, en Estados Unidos entre el 0.9% y el 3.7%; en Brasil el 2.5%; en Chile el 10% y en Argentina aproximadamente el 14%.
La edad de presentación es más frecuente entre los 20 y 40 años, pero el rango va desde los 4 a 67 años. La incidencia de VKH en niños ha aumentado en Chile en los últimos años (datos no publicados del Servicio de Oftalmología del Hospital del Salvador).
En nuestro país es más frecuente mujeres 3.5:1, similar a lo observado en Argentina y México, sin embargo Ohno en Japón refiere que el VKH es más frecuente en hombres (18).
La etiología es desconocida, pero de patogenia autoinmune, con autoinmunidad tanto humoral como celular contra los melanocitos. Ohno la ha llamado enfermedad autoinmune melanocito específica.
Las manifestaciones clínicas del VKH son:
a)Síntomas ProdrómicosEstos se presentan días o semanas previas a las manifestaciones oculares, caracterizadas por cefalea intensa a veces acompañada de nauseas y vómitos, mareos, vértigo, fiebre, tinitus, disacusia, rigidez cervical dolorosa, hipersensibilidad del cuero cabelludo. Ocasionalmente puede existir una metamorfopsia como única manifestación ocular en esta etapa.
b)Manifestaciones ocularesSe presenta como una uveítis difusa bilateral granulomatosa, en la que el paciente consulta por ojo rojo, fotofobia, epifora, dolor ocular y disminución súbita de agudeza visual, siendo generalmente de presentación.
En el segmento anterior se aprecian signos de uveítis anterior como células y proteínas en humor acuoso (tyndall celular y proteico), precipitados queráticos, nódulos sobre iris y formación de sinequias posteriores.
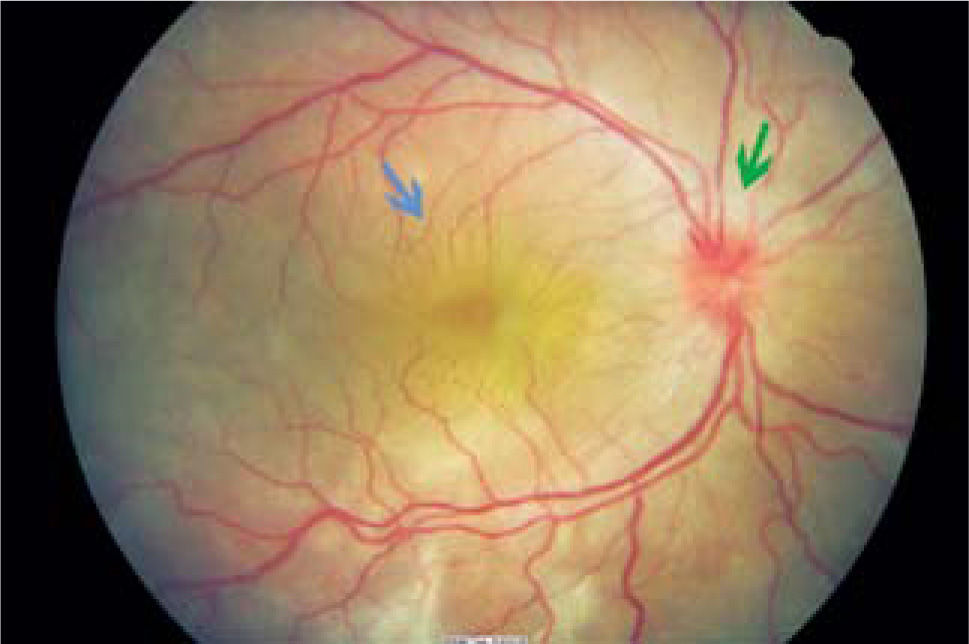
Los cambios más característicos y severos ocurren en el segmento posterior donde se observa vitreítis leve a moderada, hiperemia o edema del disco óptico y edema retinal (ver Figura 5). A medida que la inflamación aumenta, aparece un desprendimiento de retina seroso (exudativo, no regmatógeno), que es el hallazgo diagnóstico más importante. A veces pueden aparecer nódulos blanco amarillentos a nivel coriorretinal del tipo Dallen Fuchs.
, acompañado de edema de papila (flecha verde).")
Tardíamente en el curso de la enfermedad y luego de la desaparición del desprendimiento retinal, aparecen alteraciones pigmentarias de la retina, que incluye una despigmentación severa de ella (sunset glow fundus), acúmulos pigmentarios, manchas despigmentadas generalmente periféricas y fibrosis sub-retinal. Finalmente es posible ver signos de uveítis anterior crónica o recurrente.
c)Manifestaciones ExtraocularesCompromiso neurológico y auditivo: caracterizados por meningismo observado en los síntomas prodrómicos, pleocitosis del líquido cefaloraquideo, tinitus y disacusia.
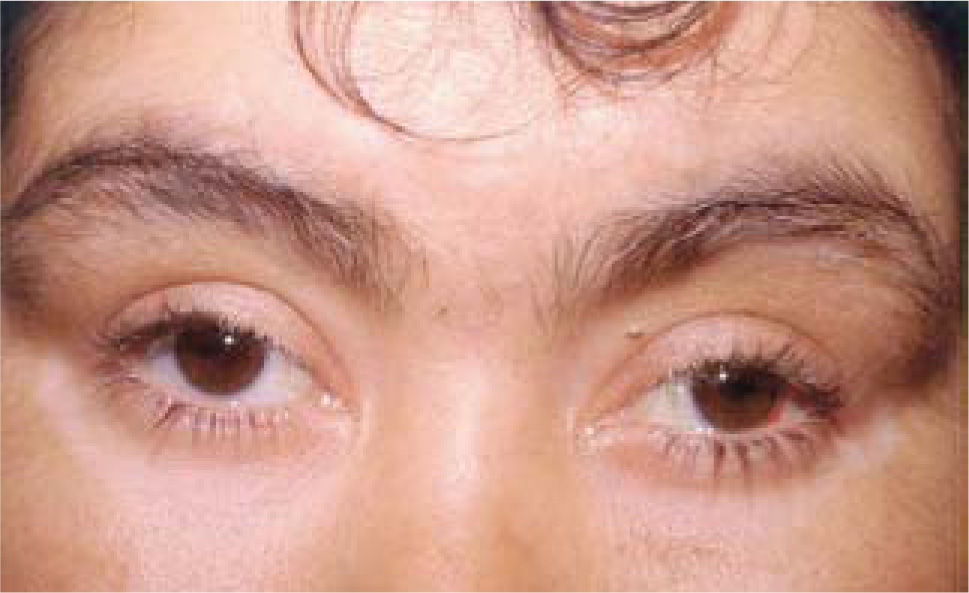
Compromiso dermatológico: como vitíligo, poliosis, alopecía y canicie; que aparecen tardíamente, habitualmente en la etapa de convalecencia de la enfermedad (ver Figura 6).
.")
El diagnóstico es eminentemente clínico, pudiendo ser de ayuda algunos exámenes complementarios como la angiografía retinal con flurosceína e indocianina verde, la tomografía de coherencia óptica, la punción lumbar, audiometría, ecografía ocular y la tipificación HLA DR4, HLA DRw53 y w15.
El tratamiento ha sido fundamental para mejorar el ominoso pronóstico que tenía el VKH. Deberá ser: precoz, sistémico, agresivo y prolongado (mínimo 8 meses a un año).
Este se realiza en base a terapia tópica con colirios midriáticos y esteroides, y eventualmente con inyecciones perioculares de corticoides de depósito. A esto se asocia un tratamiento sistémico con pulsos de metilprednisolona, para continuar con prednisona oral en dosis de 1mg/kg/día. Habitualmente se agrega un agente inmunosupresor (ciclosporina A o azatioprina). En caso de fracaso de esta terapia, están indicados agentes citotóxicos y terapia biológica, esta última con resultados alentadores en niños.
Consideraciones generales respecto al tratamientoEl enfoque terapéutico en este tipo de pacientes debe estar basado en un equipo multidisciplinario que permita realizar un diagnóstico acertado y oportuno, efectuar un tratamiento adecuado, evaluar el pronóstico de la enfermedad y la efectividad del tratamiento instaurado.
Muchas veces la tardanza en realizar un diagnóstico preciso, lleva a que estos pacientes acudan al especialista en etapas tardías y secuelares de la enfermedad, con un pronóstico mucho más reservado. Del mismo modo, un tratamiento efectivo para el compromiso de un órgano en particular, puede ser insuficiente para controlar la afección de otros órganos en una misma enfermedad.
Es importante que el médico que reciba por primera vez o maneje a este tipo de pacientes, tenga presente la posibilidad de coexistencia de enfermedad ocular en aquellos sujetos con las patologías sistémicas señaladas. La derivación oportuna al oftalmólogo ante cualquier síntoma o signo ocular, sea severo o banal, permitirá realizar un diagnóstico y tratamiento precoz (pilar en el éxito de la terapia).
Por último el tratamiento en sí puede favorecer la aparición de otras enfermedades y complicaciones tanto en el ojo, como sistémicas, que se pueden prevenir o detectar oportunamente bajo el esquema de trabajo de un equipo multidisciplinario.
Los autores declaran no tener conflictos de interés, en relación a este artículo.
El concepto de ojo rojo superficial y profundo, sólo hace mención al segmento del polo anterior que está más inflamado. En el ojo rojo superficial la mayor intensidad del ojo rojo se encuentra a nivel de los fondos de saco, y es menos intenso alrededor de la cornea, siendo un rojo rutilante. En el ojo rojo profundo, la mayor intensidad de la inflamación se encuentra en torno a la cornea, además de ser un rojo con matiz violáceo.
La escleritis corresponde a una inflamación de los vasos esclerales profundos. Se clasifica en anterior y posterior. La escleritis anterior presenta un ojo rojo profundo intenso, de color violáceo, doloroso, que puede ser difuso o nodular, que al instilar un vasocontrictor no desaparece. La escleromalacia perforans es una forma severa de escleritis anterior, caracterizada por adelgazamiento escleral en un ojo con discreta inflamación. Esta última forma se asocia en un 100% a patología sistémica.
La escleritis posterior por otro lado se caracteriza por dolor ocular a la motilidad de un ojo relativamente blanco, pero con disminución de la agudeza visual y una masa en el fondo de ojos con o sin desprendimiento de retina que refleja el proceso inflamatorio de la esclera posterior.
La epiescleritis a diferencia de la anterior, es un proceso inflamatorio de los vasos esclerales superficiales que causa un ojo rojo profundo, generalmente no son dolorosos y disminuye al instilar una gota de un vasocontrictor.