


Mucopolysaccharidosis type II (MPS II) or Hunter syndrome is an inborn error of metabolism due to lysosomal accumulation, with a recessive inheritance pattern linked to the X chromosome. It is caused by a deficiency in the activity of the lysosomal enzyme iduronate-2-sulfatase encoded by the IDS gene. Iduronate-2-sulphatase enzyme activity in plasma was measured and the IDS gene was analysed in genomic DNA by automated direct sequencing. Enzyme activity was 1.2μmol/l/h (reference value: >2μmol/l/h), while the molecular analysis detected the mutation c.1403G>A (p.R468Q), confirming the diagnosis of MPS II. In conclusion, since here in Mexico there are few groups dedicated to this family of diseases, we must emphasise the need to keep up to date and create expert teams of doctors and scientists specialised in inborn errors of metabolism.
La mucopolisacaridosis tipo II (MPS II) o síndrome de Hunter es un error innato del metabolismo por acumulación lisosomal con un patrón de herencia recesivo ligado al cromosoma X. Es causada por una deficiencia en la actividad de la enzima lisosomal iduronato-2-sulfatasa codificada por el gen IDS. Se determinó la actividad enzimática de la iduronato sulfatasa en plasma y el análisis del gen IDS en DNA genómico mediante secuenciación directa automatizada. La actividad enzimática fue de 1.2μmol/l/h (valor referencia: >2μmol/l/h) mientras que el análisis molecular detectó la mutación c.1403G>A (p.R468Q) confirmando el diagnóstico de MPSII. En conclusión, es necesario resaltar la necesidad de actualizar y crear grupos médicos y científicos especializados en errores innatos del metabolismo, ya que en nuestro país hay pocos grupos dedicados a ese grupo de padecimientos.
Mucopolysaccharidosis type II (MPS II) or Hunter syndrome is an inborn error of metabolism with a recessive inheritance pattern linked to the X chromosome which belongs to the subgroup of diseases caused by lysosomal accumulation.1,2 The primary defect is a mutation on the IDS gene which causes a deficiency in the activity of the lysosomal enzyme iduronate-2-sulfatase (I2S).1,3 It is a rare disease, with an incidence of 1.3/100,000 live births.1
The enzyme iduronate sulfatase catalyses one of the steps in the catabolism of glycosaminoglycans, resulting in accumulation of heparan and dermatan sulphate in the tissue and organs.3,4 This accumulation leads to neurological disorders, severe airway obstruction, skeletal deformity, hepatosplenomegaly and cardiomyopathy, especially mitral and aortic valve regurgitation. Differential diagnosis should be made with mucopolysaccharidosis type 1 or Hurler syndrome.5 In general, the clinical features of this disease are not apparent at birth, but occur between the ages of 2 and 4 years.1 Early symptoms of the disease include respiratory infection, umbilical and inguinal hernia, joint stiffness and dysmorphia; dysostosis and kyphoscoliosis progress rapidly, resulting in short stature. The accumulation of glycosaminoglycans in the central nervous system leads to developmental delay and generally severe intellectual disability. Airway obstruction and heart failure due to valve dysfunction are the most common causes of death, with life expectancy on average being 15 years.1,3,4,6
Management of patients with MPS requires support from a multidisciplinary medical team to deal with the systemic complications. Enzyme replacement therapy (ERT) is now available for Hunter syndrome, but as the benefits of such therapy depend on the age treatment is started, early diagnosis is of great importance.6,7 In this article, we describe the case of a patient with Hunter syndrome in whom a mutation was detected in the IDS gene.
Case reportThis was a five-year-old male patient, first pregnancy of healthy non-consanguineous parents, with a healthy two-year-old brother. Generalised hypertonia was observed at 3 months and psychomotor retardation was evident from 16 months; the consultation was for the patient's behavioural disorder characterised mainly by aggression. At the age of 3, he was diagnosed with bilateral conductive hearing loss. He had a history of repeated upper respiratory tract infections, on several occasions requiring hospital admission. He had an uncomplicated tonsillectomy at the age of 4.
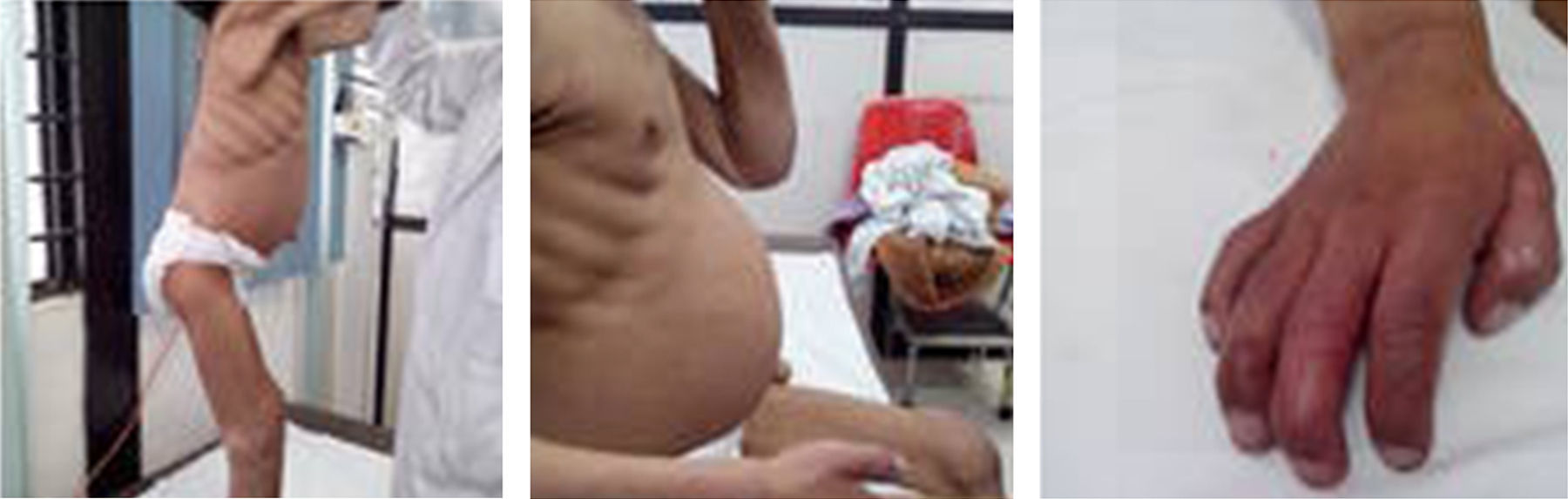
Physical examination showed the patient's general condition to be poor, with psychomotor disabilities and severe malnutrition, normocephalic, coarse facial features, prominent frontal bone, depressed nasal bridge, broad base of nose with anteverted nostrils, thick lips, prominent ears, short neck with low hairline, lung fields well-ventilated, heart sounds regular with good intensity, abdomen obese with liver 2cm below the costal margin, umbilical hernia, male genitalia, limbs with restricted extension, mainly in elbows and knees, claw hand and generalised hypertrichosis (Fig. 1).

Chest X-ray and echocardiogram within normal parameters. Abdominal ultrasound with moderate hepatomegaly, bile ducts show no structural changes. Electroencephalogram normal with no epileptic activity.
Extraction of genomic DNA from peripheral blood was carried out using conventional methods. The IDS gene was analysed by polymerase chain reaction (PCR) and direct DNA sequencing. The PCR products obtained were sequenced on an ABI 3730 Automated Sequencer (PE Biosystems, Foster City, CA).
Qualitative analysis of glycosaminoglycans (GAGs) in urine with toluidine blue positive. Iduronate sulfatase enzyme activity in plasma diminished at 1.2μmol/l/h (reference value: >2μmol/l/h), confirming the diagnosis of MPS II. Molecular analysis of the IDS gene, which codes for the enzyme iduronate sulfatase, detected that the patient was hemizygous for the mutation (c.1403G>A, p.R468Q).
The patient died of respiratory complications 6 months after being diagnosed with MPS II.
DiscussionHunter syndrome is a rare inherited disease in which, because of its progressive nature and the irreversible damage it causes, early diagnosis and therapeutic intervention are of utmost importance. In our patient, given his clinical manifestations, this syndrome could have been suspected, but over the course of his medical visits and admissions to hospital, no diagnosis was made. Recombinant human synthetic enzymes (Enzyme Replacement Therapy or ERT) are currently used as treatment for some lysosomal storage diseases, including Hunter syndrome.7
The main benefits of ERT include a significant reduction in urinary GAG excretion, with improved joint mobility, ability to walk, lung function, cardiac parameters and hearing, and reduced liver and spleen volume. However, ERT administered intravenously does not cross the blood-brain barrier, so nervous system damage cannot be repaired. Also, despite ERT, no improvement has been reported in ophthalmological or skeletal problems or respiratory function, again emphasising the importance of early diagnosis.3 ERT has been shown to be safe in patients under a year old.8 Clinical evidence shows that early interventions with ERT produce a better response to treatment with a clear improvement in patients’ quality of life.4,7 The mutation found in the patient was first described by Whitley et al. in 19939; an adenine replaces a guanine in exon 9 of the IDS gene, which produces a substitution of glutamine for arginine at position 468 on the protein. Sukegawa et al., in a study of expression, demonstrated decreased enzyme activity in fibroblasts of patients with this mutation.10 This mutation has been reported in several ethnic groups and is associated with severe phenotypes. Other mutations, R468W11 and R468L,12 have been reported in the same codon, suggesting that this site is a mutational “hot spot” for the IDS gene. Over 350 mutations have been described in the IDS gene to date.2,13
In conclusion, since here in Mexico there are few groups dedicated to this family of diseases, we must emphasise the need to keep up to date and create expert teams of doctors and scientists specialised in inborn errors of metabolism. It would be reasonable to think that the vast amount of resources that social security systems and government programmes assign to this type of disease would not only be used for medical care but also be dedicated to research and the production of new medical knowledge.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe authors declare that they have no conflict of interests.