


Sensorineural hearing loss (SNHL) is a clinically and genetically heterogeneous disease. In some populations, c.365delG mutation in the GJB2 gene represents the most frequent cause of hereditary SNHL. The great diversity of mutations in the GJB2 gene worldwide highlights the participation of ethnic background in SNHL.
ObjectiveTo describe the presence of homozygous c.del35G mutation in the GJB2 gene in a Mexican family with SNHL.
Materials and methodsA Mexican family with SNHL was included in the study. Analysis of the GJB2 gene was performed through whole exome sequencing (WES) and DNA direct sequencing analysis in all members of the family and in 100 normal controls
ResultsAffected sibs showed the homozygous c.del35G mutation in the GJB2 gene. Parents of the families were heterozygous for the molecular defect and had normal audition.
ConclusionWe describe a homozygous c.del35G mutation in the GJB2 gene through WES analysis, a homozygous mutation with a very low occurrence in Mexican population.
La pérdida auditiva neurosensorial (HNS) es una enfermedad clínica y genéticamente heterogénea. En algunas poblaciones, la mutación c.365delG en el gen GJB2 representa la causa más frecuente de la HNS hereditaria. La gran diversidad de mutaciones presentes en el gen GJB2 en todo el mundo pone de relieve la participación del genotipo étnico en HNS.
ObjetivoDescribir la presencia de la mutación homocigota c.del35G en el gen GJB2 en una familia mexicana con HNS.
Materiales y métodosUna familia mexicana con HNS se incluyó en el estudio. EL análisis del gen GJB2 se realizó a través de la secuenciación del exoma (WES) y el análisis de la secuenciación directa del DNA en todos los miembros de la familia y en 100 controles normales Resultados Los hermanos afectados presentaron la mutación c.del35G homocigótica en el gen GJB2. Los padres de los pacientes fueron heterocigotos para el defecto molecular con una audición normal.
ConclusiónSe describe la mutación homocigota c.del35G en el gen GJB2 a través del análisis de WES, una mutación homocigota observada en una muy baja incidencia en la población mexicana.
Non-syndromic sensorineural hearing loss (SNHL), a genetically heterogeneous affection, is a common hereditary disease that presents an occurrence of 1 in 1000 newborns. In relation to the onset, SNHL can be congenital, prelingual or postlingual. All types of inherited patterns have been observed in SNHL (autosomal dominant, autosomal recessive, mitochondrial and X-linked) being the autosomal recessive form the most frequently observed.1 The syndromic form of sensorineural hearing loss is defined when other systemic manifestations are present; many syndromes with the presence of hearing loss are described in the OMIM (Online Mendelian Inheritance in Man). In some cases, differential diagnosis between syndromic and non-syndromic hearing loss is not easy to perform even with molecular studies because both types of hearing loss can be due to affections of the same gene.
It is considered that 200–250 genes are involved in SNHL.2 Recently, more than 80 genes, around 1000 mutations, and 140 loci have been associated with SNHL (http://hereditaryhearingloss.org/).
Mutations in the GJB2 gene are a frequent etiology of hereditary SNHL, they represent about 50% of such cases in several populations.3–5 Patients that carrier homozygous mutations in the GJB2 gene harbor a wide spectrum of hearing loss that ranges from moderate to profound NSHL; this data supports the influence of epigenetic and environmental factors in the phenotypic manifestations.6 Previous studies show a rare presence of homozygous mutations in the GJB2 gene in Mexican population.7,8
The GJB2 gene encodes the gap junction beta-2 protein connexin 26; six connexin molecules form connexons between adjacent cells, responsible for the exchange of ions and molecules among contiguous cells.9 Connexins are present in connective and epithelial tissues of the cochlea, which primordial function is to maintain the normal audition.10
Improvement of the technology plays an important role in the diagnosis of different pathologies, whole exome sequencing (WES) represents an important tool in the identification of the genetic etiology in individuals with SNHL. In the present study we describe a family with SNHL and homozygous mutation in the GJB2 gene detected by WES.
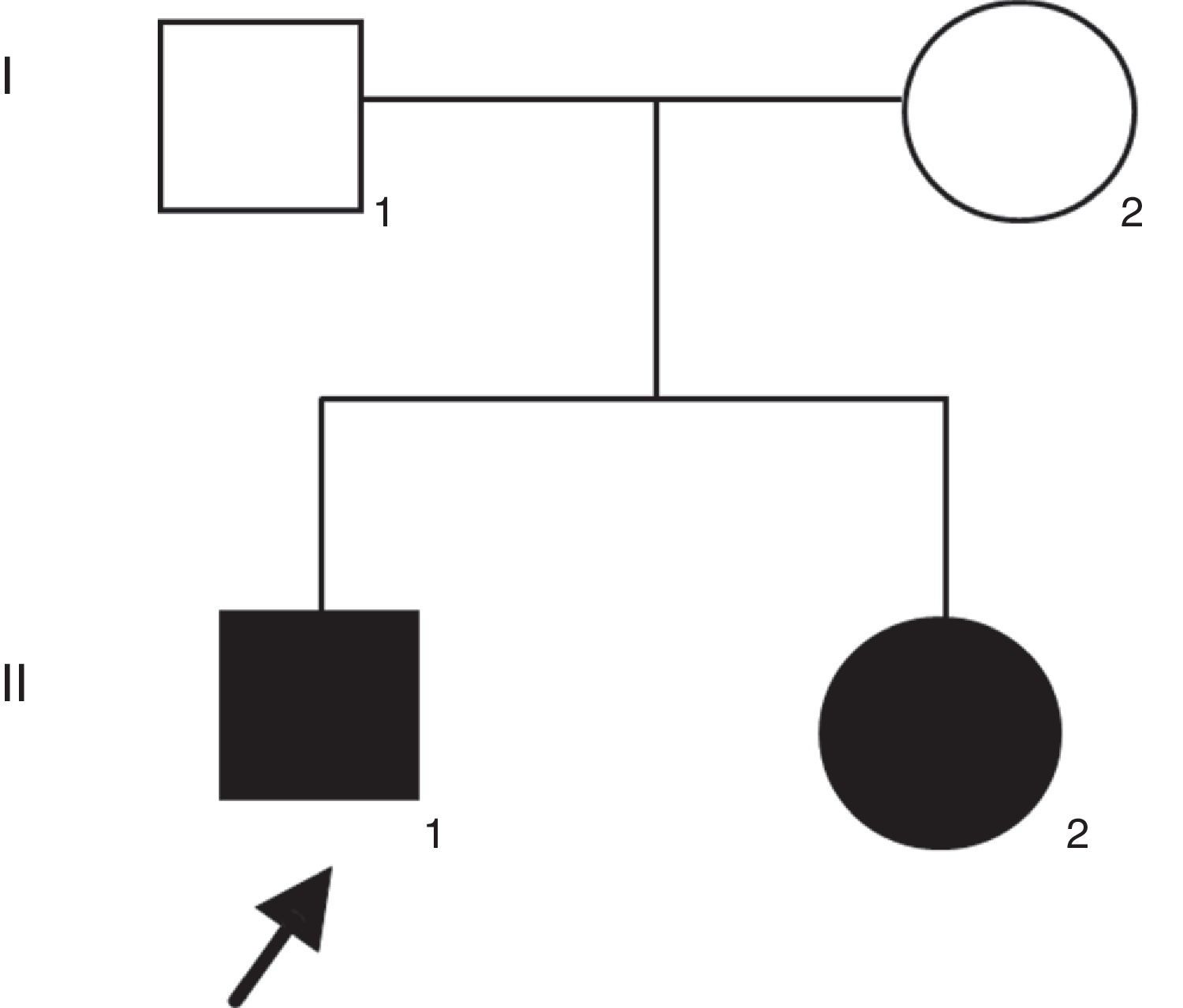
Patient and methodsThe proband was a 21-years-old man with profound congenital deafness in both ears. He was the first product of healthy and non-consanguineous parents. He has a 19-years-old sister with congenital deafness in both ears, as well (Figure 1). Family history was negative for intellectual disability or congenital malformations. No history of prenatal exposure to teratogens, maternal illness or use of aminoglycoside antibiotics was recorded. His mother had an uneventful pregnancy with a vaginal delivery at 40 weeks of gestation. His psychomotor development was normal. There was no evidence of syndromic deafness. Computed tomography of the skull showed normal structures. Thyroid parameters were normal. Informed consent was obtained from the patients to participate in the study.
Whole exome sequencing (WES)
Two μg of genomic DNA was submitted to Axeq Technologies for human exome capture using SureSelect human All Exon kit (Agilent Technologies). Axeq Technologies performed sample validation, library preparation, exon enrichment, clustering and sequencing using illumina HiSeq 2000 Sequencer. Approximately, 63,000,000 reads of an average size of 107bp per sample were returned to us as two fastq files (one file per orientation). Each pair of fastq files were aligned to the human genome (hg19) using Novoalign (http://www.novocraft.com). Common variants were removed using allele frequency information from the NHLBI ESP (http://evs.gs.washington.edu/EVS). The VCF files were annotated using snpEff (snpeff.sourceforge.net).
Mutation analysisTo confirm the WES findings, DNA sequencing analysis of the GJB2 gene was performed through Sanger method. The coding exon and flanking intronic regions of the GJB2 gene were amplified by PCR. All the PCR products were directly sequenced. DNA was analyzed on an ABI 3730XL automated sequencer (Applied Biosystems, Inc., Foster City, CA, USA). The GJB2 gene was sequenced in all family members and in 100 normal controls to discard polymorphisms.
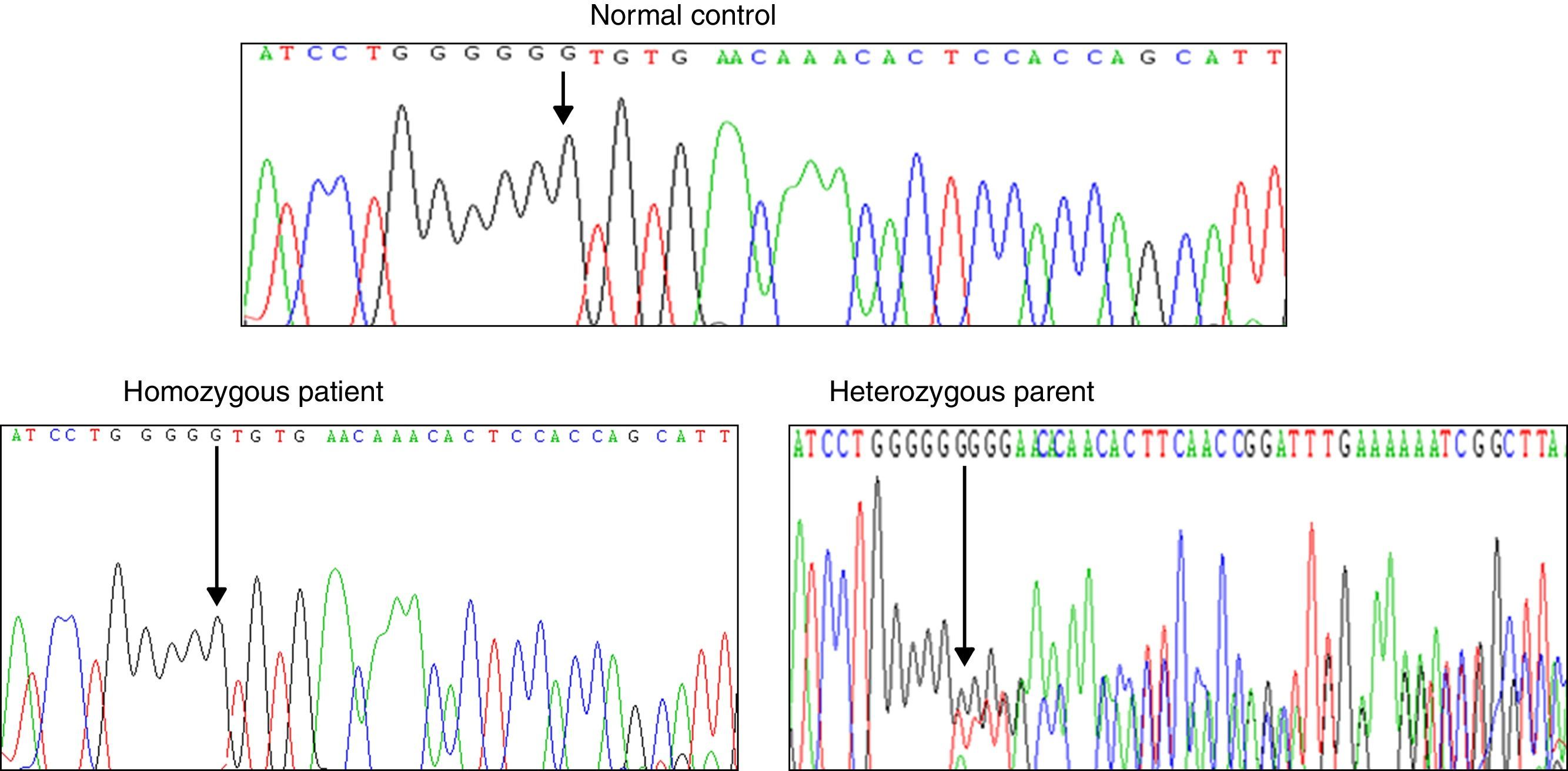
Results and discussionFrom the initial raw variant file (vcf) contained more than 150,000 putative variants, we isolated the high quality, non-synonymous putative coding variants with our quality filter in conjunction with the annotation tags generated by snpEff (Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92.). After exploring potential genes involved in NSHL, we found the homozygous mutation c.del35G in GJB2. This finding was confirmed through Sanger method (Fig. 2). Audiology studies revealed profound neurosensorial hypoacusia in the affected sibs of the family.
 in the proband (homozygous), parents (heterozygous) and normal control.")
Recently, several studies have been performed in the GJB2 gene that encodes Cx26 connexin. GJB2 mutations produce several degrees of hearing impairment even in the same family, demonstrating the clinical heterogeneity observed in SNHL. The genetic analysis of the GJB2 gene (WES and DNA sequencing) in our family with SNHL identified the homozygous c.35delG mutation, this molecular affection was present in the sibs and absent in the normal controls. Both parents were heterozygous for this molecular defect.
Whereas previous studies reveal a very low frequency of this type of mutation in Mexican population, this molecular affection is the most prevalent GJB2 mutation in Caucasian population.7,8,11 Conversely, analysis of the prevalence of GJB2 mutations in eastern populations shows that c.235delC and not c.35delG is the most frequent cause of SNHL. This difference in the prevalence of the types of mutations in the GJB2 gene indicates the influence of ethnic background.
The complexity of the genetic component in SNHL is denoted by the great quantity of genes involved in the pathogenesis of the disease. Currently, more than 80 genes have been associated to SNHL (http://hereditaryhearingloss.org/), which remarks the difficult to identify the molecular alteration in these patients. Even more, the great variability of the gene affections in each population makes no possible to stablish a specific panel to identify the gene and the mutation involved in the molecular etiology of the SNHL.
In conclusion, we describe a Mexican family with SNHL and homozygous mutation c.35delG in the GJB2 gene. The new techniques of the genomics era (such as whole genome sequencing, whole exome sequencing or targeted next generation sequencing) have an important role in research or diagnostic areas; they represent excellent tools in the detection of molecular etiology as in SNHL due to its high genetic and clinical heterogeneity.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe authors declare that they have no conflict of interests.