


La enfermedad de Hartnup es un trastorno congénito del metabolismo, causado por una mutación en el gen SLC6A19 el cual codifica para un transportador de aminoácidos neutros presente en el intestino y los riñones. El cuadro clínico característico presenta ataxia cerebelosa, dermatitis pelagroide relacionada a fotosensibilidad, aminoaciduria y síntomas neuropsiquiátricos, comúnmente depresión, irritabilidad e insomnio. El cuadro clínico es atribuido a la ineficiente producción de nicotinamida y a las alteraciones en la síntesis del neurotransmisor 5–hidroxitriptamina (5-HT) causadas por la disminución en la absorción de su precursor el L-triptófano (W). Ésta revisión pretende ejemplificar la relación directa entre la disrupción en el metabolismo de un nutriente elemental y la compleja presentación clínica que deriva de la misma.
Hartnup's disease is an inherited disorder of metabolism caused by a mutation in the SLC6A19 gene which encodes for a neutral amino acid transporter present in the intestine and kidneys. The characteristic clinical picture of the disease is cerebellar ataxia, pellagra like skin rash related to photosensitivity, aminoaciduria and neuropsychiatric symptoms, commonly depression, irritability and insomnia. The clinical picture is attributed to the inefficient production of nicotinamide and alterations in the synthesis of the neurotransmitter 5-hydroxytryptamine (5-HT) caused by decreased absorption of its precursor L-tryptophan (W). The aim of this review is to illustrate the direct link between the disruption in the metabolism of a nutrient elemental and the complex clinical presentation that derives from it.
La enfermedad de Hartnup (EH) es la patología más común relacionada con errores congénitos en el metabolismo de aminoácidos, recibe su nombre en honor a la familia Hartnup, originaria de Londres donde fue descrita por primera vez en 1956 por Baron et al.1, quienes en su manuscrito publicado en la célebre revista Lancet, la definen como: “Una dermatosis similar a la pelagra que cursa con ataxia cerebelosa, aminoaciduria constante y otras manifestaciones bioquímicas bizarras de transmisión autosómica recesiva”.
La pelagra (pelle: piel, agra: áspera) una enfermedad causada por la deficiencia dietética de vitamina B3 o niacina, caracterizada por la tríada clásica de: dermatosis, diarrea y demencia, no explicaba la aminoaciduria y la ataxia que presentaban ocho miembros de la familia Hartnup, la cual parecía consumir una dieta adecuada en contenido de vitaminas del complejo B. Baron et al.1, apoyados en la similitud entre la dermatosis propia de la EH y la pelagra creyeron que el defecto bioquímico inmediato se encontraba en el metabolismo del ácido nicotínico, un derivado de la niacina.
En 1960, los estudios de concentración de aminoácidos en plasma, orina y heces realizados por Milne et al.2, en pacientes no emparentados con los descritos originalmente por Baron pero con las mismas características clínicas, sugirieron que el defecto bioquímico se encontraba en “un transportador luminal de aminoácidos localizado en el intestino y el riñón o bien en un defecto en la conversión de triptófano hacia ácido nicotínico”. En el 2004 gracias a la secuenciación del genoma humano se postula un gen candidato cuya mutación explicaría las alteraciones bioquímicas características de la EH, se trata del gen SLC6A19 (solute carrier family 6 member 19) cuyo locus corresponde a 5p15.33 y el cual codifica para un transportador epitelial de aminoácidos neutros dependiente de Na+ e independiente de Cl-, el cual se expresa en el borde en cepillo de los enterocitos y en el epitelio de recubrimiento del túbulo renal proximal3.
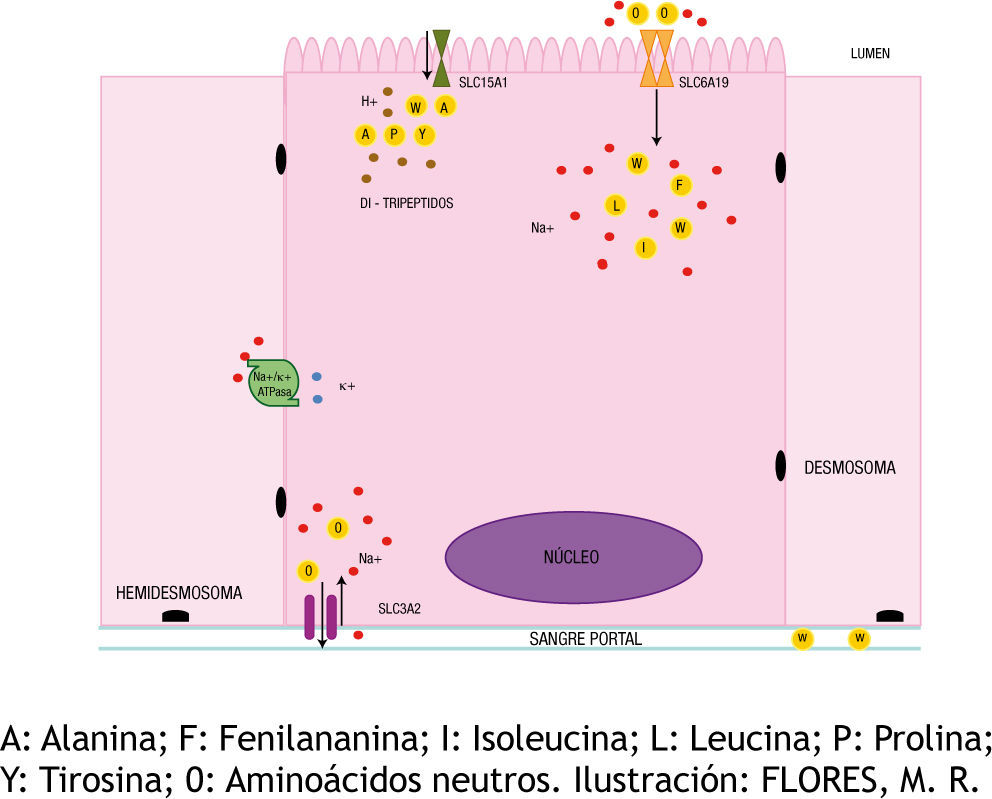
FisiopatologíaEl L-triptófano (W, por su código de una letra) no puede ser sintetizado directamente por el ser humano lo cual lo convierte en un aminoácido esencial que debe ser consumido en la dieta, con un requerimiento estimado de 7mg/kg/día. Una vez que las proteínas de la dieta son ingeridas, la actividad de las enzimas digestivas como la pepsina en el estómago y la quimiotripsina liberada desde el páncreas, desdoblan las mismas hasta aminoácidos libres, dipéptidos, tripéptidos y polipéptidos de cadena corta, estos tres últimos son susceptibles de ser hidrolizados por las peptidasas que se encuentran en las microvellosidades de la membrana luminal de los enterocitos antes de ser absorbidos hacia el torrente sanguíneo.
El transportador de aminoácidos neutros SLC6A19 es una proteína con 12 dominios transmembrana cuyos extremos -NH2 y –COOH tienen una localización citoplásmica. La absorción de W y otros aminoácidos neutros desde el lumen intestinal hacia el citoplasma de los enterocitos es Na+ dependiente, lo cual significa que utiliza un mecanismo de trasporte activo secundario, con el gasto de energía concomitante, derivado del consumo de ATP por la NA+/K+ ATPasa. Para que los aminoácidos neutros pasen del compartimento citoplasmico de los enterocitos hacia la sangre portal deben atravesar la membrana basal a través del transportador SLC3A2 mediante antiporte con Na+. En la EH, el gen SLC6A19 se encuentra mutado, se han descrito al menos seis puntos de mutación en pacientes con dicha patología4. La escasa absorción de W en estos pacientes es explicada por la existencia del transportador SLC15A1 el cual es capaz de absorber dipéptidos y tripéptidos que contienen triptófano y otros aminoácidos neutros a través de simporte con H+ (fig. 1).

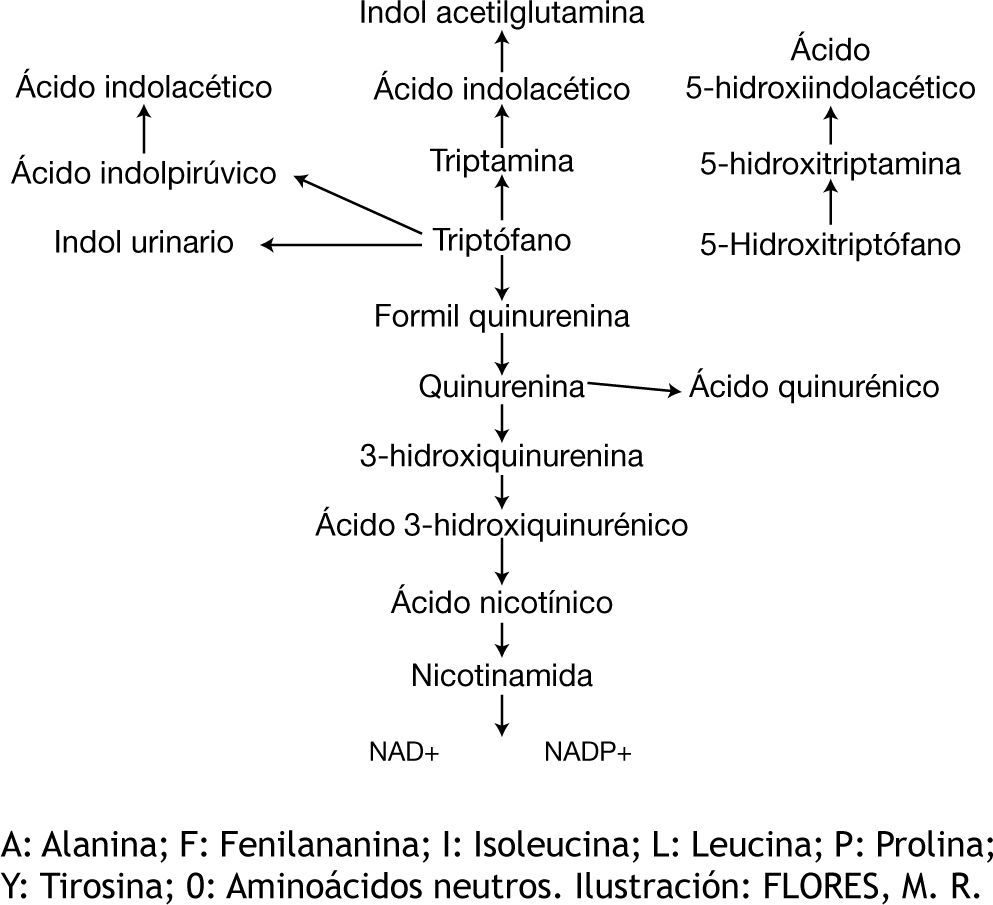
El L-triptófano es metabolizado a través de la vía de la quinurenina hacia nicotinamida componente fundamental de las moléculas NAD+ (nicotinamida adenina dinucleótido) y NADP+ (nicotinamida adenina dinucleótido fosfato) que a su vez participan en importantes reacciones de óxido-reducción en el metabolismo celular. La enzima triptófano 2-3 oxigenasa es la enzima limitante de este proceso5 (fig. 2). Las exigencias metabólicas propias de las células de la epidermis debidas en parte a su tasa de mitosis han llevado a postular que un déficit en la producción de NAD+ y NADP+ podría explicar la aparición de áreas de hiperqueratosis predominantes en zonas expuestas a luz solar, las cuales asemejan la dermatosis por fotosensibilidad similar a pelagra de la descripción clásica de la enfermedad.

La biosíntesis de 5-hidroxitriptamina (5-HT) o serotonina y melatonina a partir de L-triptófano por parte de las neuronas en el sistema nervioso central parece explicar la ataxia cerebelosa y los cuadros de depresión e insomnio en los pacientes con EH. En primer lugar la escasa absorción de W a través de SLC15A1 restringe las concentraciones de éste aminoácido que traspasan la barrera hematoencefálica. El W es absorbido por las neuronas serotoninérgicas a través de los transportadores SLC3A2 y SLC3A7. Ya en el citoplasma neuronal, es convertido en 5-hidroxitriptófano por acción de la triptófano hidroxilasa, después de sufrir descarboxilación por medio de la enzima descarboxilasa de aminoácidos neutros, el 5-hidroxitriptófano se transforma en el neurotransmisor 5-HT. La proyección neuroanatómica de las vías serotoninérgicas desde el núcleo dorsal del rafe, el área tegmental ventral y la médula espinal hacia el cerebelo explicaría la ataxia cerebelosa de los pacientes con EH que se manifiesta como la descoordinación de los movimientos voluntarios. Mientras que la depresión puede asociarse a la importante vía serotoninérgica encontrada en el sistema mesolímbico, una estructura filogenéticamente conservada del SNC que ha sido implicada en el control de las emociones en el ser humano.
En segundo lugar, la 5-HT es convertida a través de dos reacciones enzimáticas en melatonina, molécula que ha sido asociada a la inducción del sueño. En la glándula pineal localizada en el techo del diencéfalo, la 5-HT es captada por los pinealocitos en presencia de oscuridad, mientras que la N-acetilasa de 5-HT y la O-metiltrasferasa del hidroxiindol producen melatonina, la cual difunde a través del torrente sanguíneo hasta alcanzar los receptores MT1 y MT2 en la sustancia reticular del tallo encefálico donde inducen al sueño. De ésta manera, se observa claramente la importancia del W en la cronobiología y el ritmo circadiano en el ser humano6.
La aminoaciduria presente en la EH está dada por la excreción no solo de W sino de otros aminoácidos neutros no polares como isoleucina, leucina, glicina, prolina, valina, fenilananina etc. En los riñones, las células del túbulo contorneado proximal tienen como principal función la reabsorción de los nutrientes filtrados desde el glomérulo para su reaprovechamiento por el resto de la economía7. En los pacientes con EH las mutaciones en SLC6A19 impiden la correcta reabsorción de los aminoácidos neutros que ingresaron al torrente sanguíneo por medio del transportador SLC15A1 localizado en el borde en cepillo luminal de los enterocitos. Este defecto permite una concentración urinaria alta de aminoácidos neutros8, uno de los criterios diagnósticos de la EH.
DiagnósticoHistóricamente, el diagnóstico de la EH ha sido clínico y bioquímico. Como se mencionó con anterioridad, la dermatosis pelagroide está explicada por la deficiencia relativa de NAD+ y NADP+ en las células de la epidermis, por otro lado, las alteraciones neuropsiquiátricas como la ataxia, depresión e insomnio por la escasez de serotonina y melatonina en el SNC. Lo anterior, así como la detección de aminoácidos neutros en una muestra de orina de 24 hora sugieren altamente la presencia de esta enfermedad. Actualmente, la detección de concentraciones anormalmente disminuidas de ácido 5-hidroxiindolacetico, un metabolito originado por la degradación de la serotonina a través de la monoamino oxidasa B (MAO-B), es detectado en orina a través espectrofotometría utilizando como colorante el 1-nitroso, 2-naftol, permitiendo de ésta forma, un diagnóstico bioquímico de certeza (concentración urinaria de ácido 5-hidroxiindolacetico <3mg/24 horas)9. El diagnóstico a través de genética molecular puede realizarse mediante el empleo de sondas complementarias a la secuencia del gen SLC6A9 mediante el método de hibridación fluorescente in situ (FISH) reservando la secuenciación o amplificación del gen para estudios de investigación.
TratamientoEl conocimiento de la etiología y fisiopatología de la EH permite deducir el tratamiento racional de la misma. En primer lugar, estos pacientes se beneficiarán de recibir alimentos complejos que sean ricos en aminoácidos neutros, no se recomiendan los complementos elementales, puesto que la absorción intestinal en pacientes con EH es a través de dipéptidos y tripéptidos aprovechando el transportador SLC15A1. De esta manera se espera aumentar la concentración de W que es metabolizada hasta NAD+ y NADP+ para paliar la dermatosis pelagroide. Los síntomas neuropsiquiátricos pueden presentar mejoría con el uso de inhibidores selectivos de la recaptura de serotonina, puesto que estos aumentan la vida media de dicho neurotransmisor. El uso de ramelteón un agonista de los receptores MT1 y MT2 parece disminuir el insomnio relacionado a la baja concentración de melatonina que alcanza estos receptores en el núcleo supraóptico y a lo largo de la sustancia reticular10. Esta breve revisión nos muestra la importancia en el conocimiento de la bioquímica y la fisiopatología que llevan a la disrup - ción en el metabolismo de un nutrimento esencial como el W para poder entender la complejidad de la presentación clínica de una enfermedad que a pesar de ser poco común (incidencia estimada de 1:24000 recién nacidos vivos) nos ejemplifica la importancia de la enseñanza de las ciencias básicas en la medicina del siglo XXI.
FinanciamientoNo se recibió financiamiento para éste estudio.
Conflicto de interésEl autor no tiene ningún interés financiero en competencia para declarar.