


Introducción: La distrofia muscular de Duchenne (DMD) está caracterizada por debilidad muscular, alteraciones de la retina y sinapsis de las neuronas corticales.
Objetivo: Determinar las alteraciones oftalmológicas en pacientes con DMD.
Métodos: Estudio observacional, prospectivo y transversal, se incluyeron pacientes con diagnóstico de DMD bajo un programa de rehabilitación en México, fueron sometidos a exploración oftalmológica y est rabológica que incluyó: valoración de agudeza visual, esquiascopía, capacidad visual, agudeza estereoscópica, percepción de los colores, determinación de tipo y magnitud de estrabismo, movimientos de seguimiento y sacádicos. Analizados con estadística descriptiva.
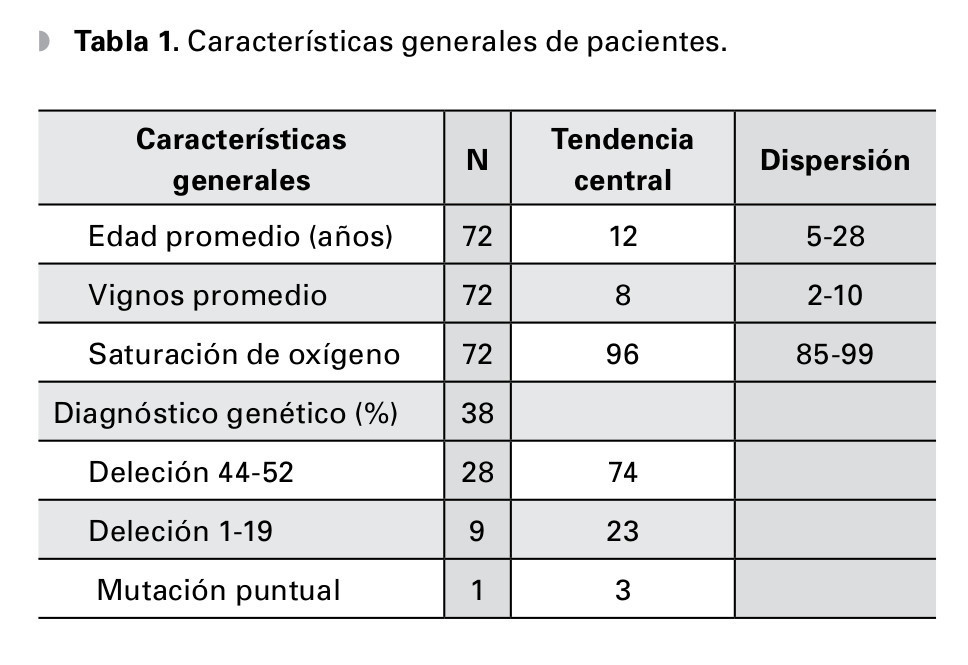
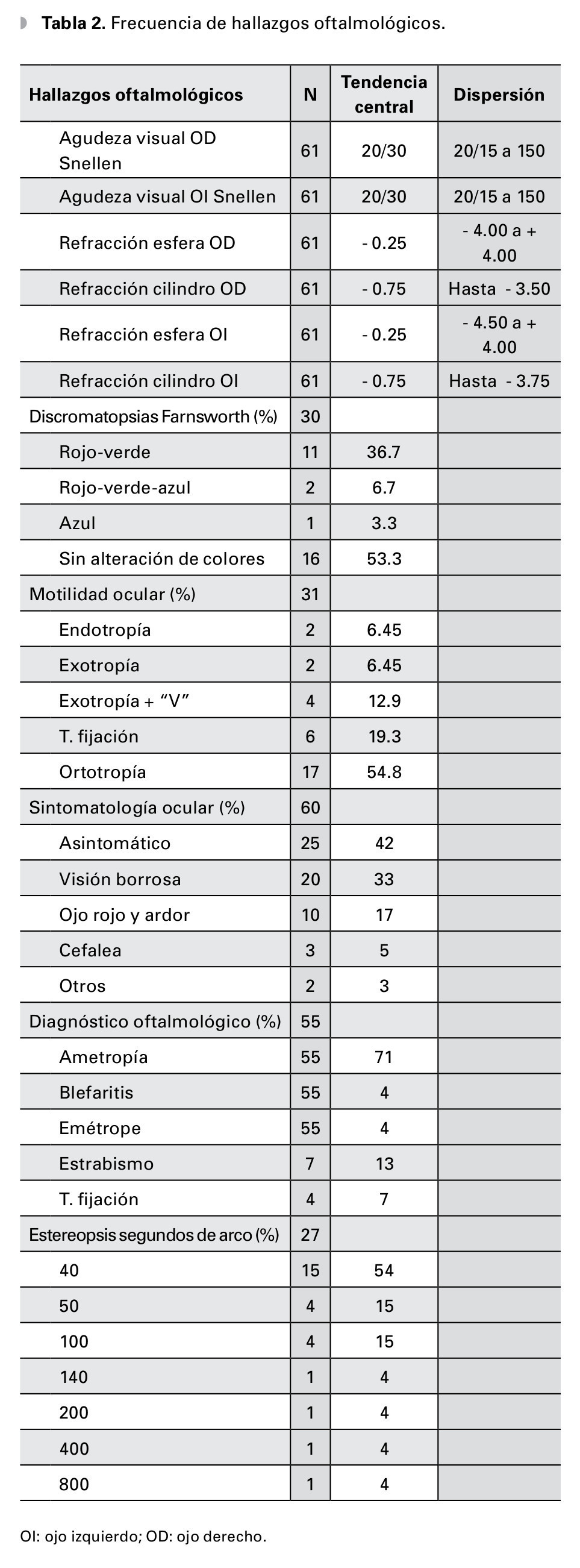
Resultados: Setenta y dos pacientes valorados, edad: 13 ± 5 años y valoración clínica general con Vignos 8 (rango 2 a 10); 38 pacientes con diagnóstico molecular confirmado, 74% encontrados con deleción de exones 44 al 52, 23% del 1 al 19 y 3% mutación puntual. Al examen oftalmológico: la agudeza visual de 0.2 LogMAR en ambos ojos, el equivalente esférico general fue de -0.50, siendo el defecto más frecuente el astigmatismo miópico simple en el 85% de los casos. La estereoagudeza fue de 93 ± 146 segundos de arco, las discromatopsias revelaron: 36.7% para rojo-verde, 6.7% rojo-verde-azul y 3.3% azul, el 45% tuvo algún trastorno de la motilidad ocular, tal como endo o exotropía en 25.8% y trastorno de fijación en 19.3%.
Conclusiones: Las manifestaciones oftalmológicas en DMD sugieren: trastornos en la percepción del color rojo-verde, deterioro del control cerebral de la fijación y la atención, involucro de los músculos extraoculares y pérdida de estereopsis, los pacientes con deleción de exones del 44 al 52, desarrollan las alteraciones más severas, relacionadas con mayor edad y progresión de la enfermedad.
Introduction: Duchenne Muscular Dystrophy (DMD) is characterized by muscle weakness and synaptic disorders in both retinal and cortical neurons.
Objective: To determine ophthalmologic disorders in patients with DMD under rehabilitation.
Methods: Observational, prospective and transversal study. Patients with DMD diagnosis under a rehabilitation program in Mexico underwent full ophthalmologic exploration by the same examiner, including: visual acuity, refraction, best corrected visual acuity, stereoscopic acuity, chromatic vision test, strabismus type and magnitude and eye slow-fast motility evaluation. Data was analyzed with descriptive statistics.
Results: Seventy-two patients were included, their average age was 12 (range5-28) and their average Vignos score was 8 (range 2 to 10). Molecular diagnosis was available for 38 patients, 28 had deletion of exons 44 to 52 (74%), 9 had deletion of exons 1 to 19 (23%) and one had a point mutation. Mean visual acuity was -0.2 logMAR, Visual symptoms were present in 58%, but clinical findings included small ametropias in 71% (mean sphere -0.25, mean cylinder -0.75D), dyschromatopsia in 47% (35% red-green, 8% red-green-blue and 4% blue), steroacuity under 40 degrees in 46% and motility disorders in 45% (25.8% eso or exotropy, 19.3% fixation disorders).
Conclusions: DMD is frequently seen with ophthalmologic findings that suggest an alteration of photoreceptor neuronal conduction, a deterioration of extraocular muscle central control and a subsequent loss of stereopsis. Ophthalmologic manifestations of DMD can include: Red-green color perception alterations, deterioration of fixation and attention due to poor brain control, extraocular muscle alterations and stereopsis loss. Patients with deletion of exons 44 to 52 develop the most severe alterations, related with age and disease progression.
• Introducción
La distrofia muscular de Duchenne (DMD) descrita en 1868,1 es la más común y grave de las distrofias musculares progresivas, con una incidencia de uno por cada 3 500 varones nacidos vivos,2 su carácter hereditario es recesivo ligado al sexo; la progresión de la enfermedad se caracteriza por deterioro de las fibras musculares y consecuentemente disminución de la fuerza muscular, que puede conducir a invalidez dentro de los primeros 10 años y provocar la muerte por insuficiencia respiratoria o cardiaca en la segunda década de la vida.3,4
El gen anormal responsable (gen DMD) está localizado en la región 21 del brazo corto del cromosoma x (Xp 21), contiene 79 exones que codifican para la síntesis de proteína distrofina y sus isoformas, las cuales están presentes en diferentes tejidos del organismo. La distrofina es una proteína que evita el daño de la membrana de las células musculares durante la contracción, se expresa en el músculo estriado esquelético, cardiaco y liso, se describen otras localizaciones como el sistema nervioso, incluyendo la retina, las células de Purkinje, paredes vasculares cerebrales como barrera hemato-encefálica y en las sinapsis de las neuronas corticales.5 Para su diagnóstico se han descrito mutaciones en el gen responsable: deleciones que representan del 65% al 72% de los casos, las duplicaciones del 5% al 7% y las mutaciones puntuales del 20% al 30%.6-8
Dentro de las manifestaciones clínicas, se observa pérdida de la capacitad de ambulación, deformidades de la columna, hipoventilación, enfermedades respiratorias, cardiacas, trastornos de la motilidad del tubo digestivo, desnutrición y trastornos de la visión.9,10 La dificultad que conlleva el estudio de estos pacientes es claro, ante la escasez de las publicaciones respecto a las funciones visuales. El electrorretinograma resulta uno de los estudios útiles para diagnóstico complementario en apoyo de diversas variables de la enfermedad, de manera que se pueden identificar tres isofomas diferentes de distrofina (Dp427, Dp260 y Dp71), las cuales se relacionan con la gravedad del padecimiento,11 se han reportado trastornos de la neurotransmisión a nivel de fotorreceptores y sinapsis excitatoria de la célula ON-bipolar determinado por electrorretinograma en un estudio de 11 niños,12,13 alteraciones en la percepción de los colores rojo-verde,14 alteraciones del campo visual encontrados en niños sometidos a campimetría computada y atribuidos a pérdida de la fijación del campo de ambos ojos hasta 10/11XX.15 En niños con DMD sometidos a electrooculografía (EOG), se demostró que las sacadas fueron significativamente más largas y más lentas en el grupo de DMD con respecto al grupo control.16 Poco se ha estudiado sobre la relación de esta enfermedad con estrabismo y otros trastornos de la motilidad ocular.
El objetivo del presente artículo es determinar las alteraciones oftalmológicas en pacientes con DMD.
• Métodos
Estudio observacional descriptivo, prospectivo y transversal analítico. Estudio realizado en el Centro de Rehabilitación Infantil Teletón de Occidente (CRIT), con autorización de la comisión de investigación del Centro, cumpliendo los lineamientos de la declaración de Helsinki y previa firma de carta de consentimiento informado. El estudio fue realizado del 01 de enero de 2000 al 01 de enero de 2012, con pacientes diagnosticados con DMD, sin selección de edad. Todos los pacientes fueron clasificados con escala de Vignos, que valora la funcionalidad y progresión clínica de la enfermedad.17 Una vez ingresados al estudio se determinaron causas de abandono del mismo, incluyendo defunción o ausencia por decisión propia.
Todos los pacientes con diagnóstico clínico, familiar o molecular confirmado de DMD, se ingresaron a un programa de rehabilitación. Se realizó exploración oftalmológica y estrabológica que incluyó agudeza visual, la cual fue obtenida con optotipos de Snellen y cuantificada según lo aceptado por la Organización Mundial de la Salud; esquiascopía, que en menores de 10 años de edad fue realizada bajo cicloplejía, aplicándose ciclopentolato al 1% y en niños mayores de 10 años, se realizó sin cicloplejía. Se obtuvo la capacidad visual con corrección, se realizó biomicroscopía y fundoscopía. Se registró la agudeza estereoscópica por medio de la prueba de Titmus "mariposa" a 40 cm con la corrección óptica, de acuerdo a diagnósticos de ametropía, siendo cuantificada en segundos de arco. Se aplicó test de Lang para corroborar las funciones estereoscópicas. El estudio de la percepción de colores fue realizado con el test de Farnsworth de 16 tonalidades, bajo una luminosidad de 329 lux, aplicando la clasificación de Varriest para discromatopsias adquiridas.18 La exploración estrabológica fue realizada determinando el tipo y magnitud de estrabismo en dioptrías prismáticas, dinámica, oculomotora, movimientos de seguimiento y sacádicos. Finalmente, los resultados fueron aplicados a estadística descriptiva. Las frecuencias de las características de los pacientes se presentaron en media o mediana, desviación estándar y proporciones o rangos. Se hizo comparación de la distribución de promedios en las variables dependientes numéricas de las manifestaciones oftalmológicas de la DMD, con los grupos de mutación genética con ANOVA. Se usó SPSS versión 19.
• Resultados
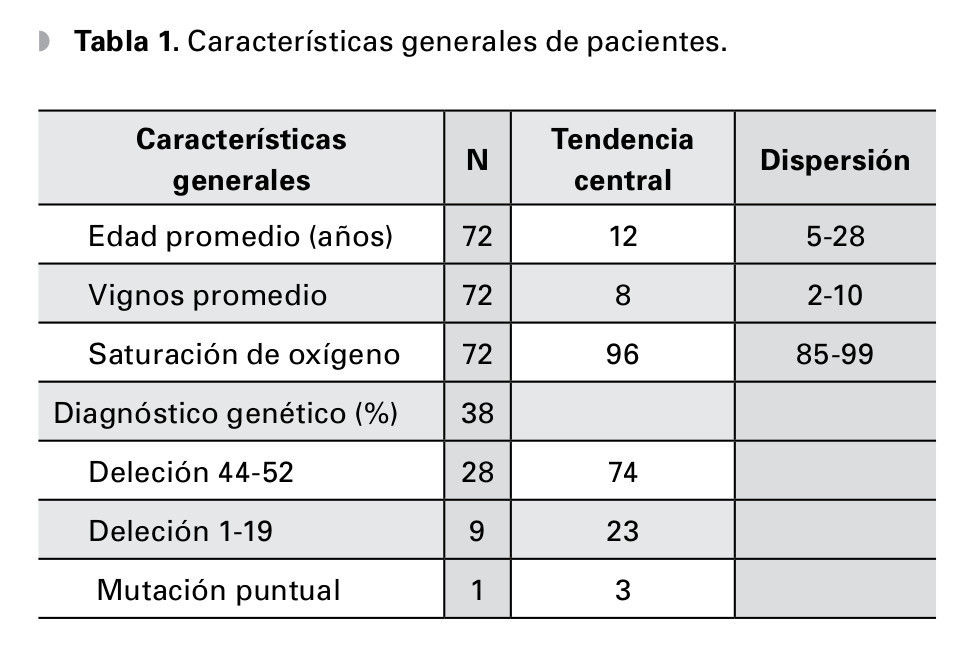
Fueron estudiados 72 pacientes, edad 13 ± 5 años, rango 5 a 28 años de edad, la valoración clínica general registró un nivel avanzado de progresión de la enfermedad con Vignos de 8 (rango 2 a 10); se pudo obtener el estudio molecular en el 53% de los casos, demostrando como causas de la enfermedad las deleciones en el 90%, lo cual la determina como la mutación más frecuente; los grupos de exones más afectados fueron las regiones del 44 al 52 y del 1 al 19, sólo hubo una mutación puntual (Tabla 1).
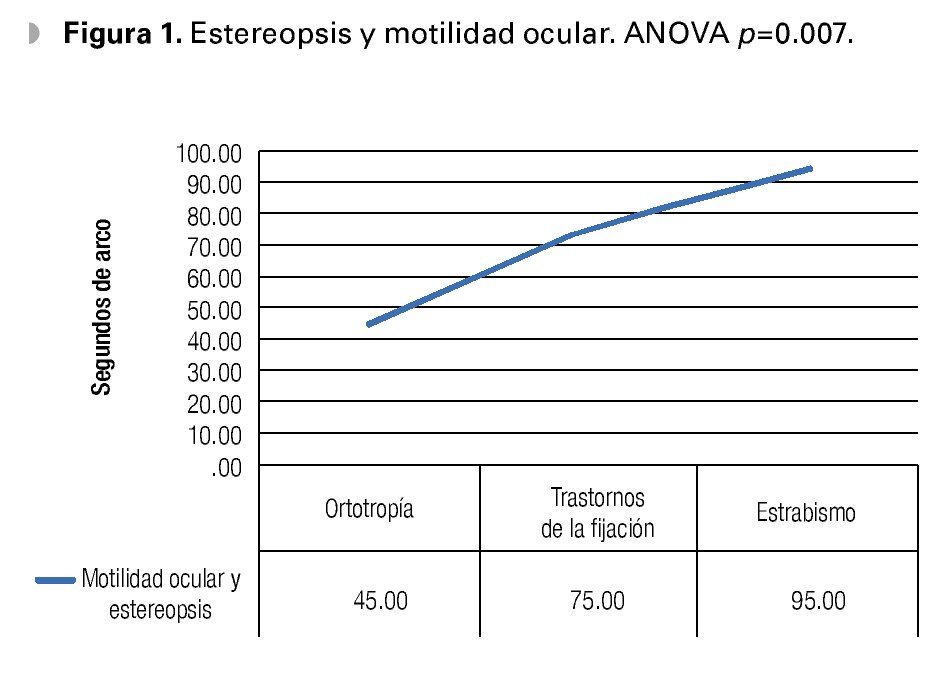
En el estudio clínico la agudeza visual mostró ser de 20/30 en promedio, con rango de 20/15 a 20/150 para ambos ojos, el equivalente esférico general fue de -0.50, siendo el defecto refractivo más frecuente el astigmatismo miópico simple en el 85% de los casos; la estereoagudeza general fue de 93 ± 146 segundos de arco. Los pacientes con trastornos de la fijación tuvieron 75 segundos de arco, y los pacientes con estrabismo tuvieron 95 segundos de arco (Figura 1).
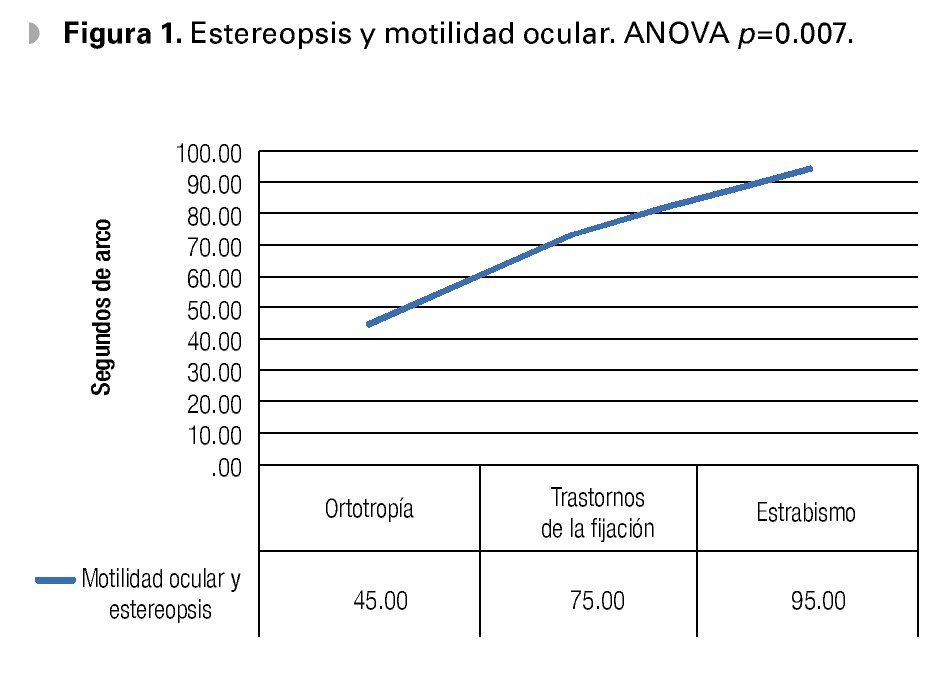
•Figura 1. Estereopsis y motilidad ocular. ANOVA p=0.007.
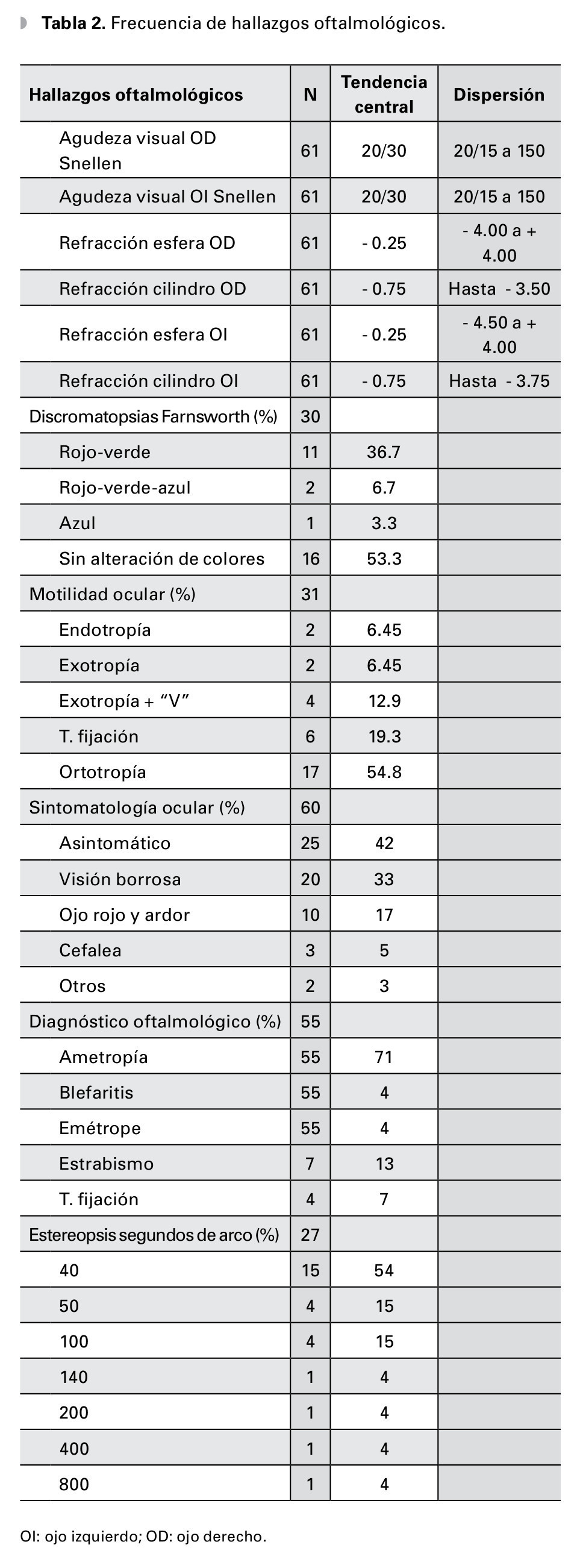
Las discromatopsias más frecuentes encontradas fueron: protan más deuteranomalía 11 casos, seguidos de protan-deuteran + tritanomalía dos casos y sólo un paciente presentó tritanomalía pura. Los trastornos de la motilidad ocular fueron endotropías n=2, exotropía comitante n=2, exotropía con estrabismo vertical n=2 y dificultad para mantener la fijación n=6; y ortotropía n=17. Los diagnósticos oftalmológicos más frecuentes asociados fueron ametropías, estrabismo y trastornos de la fijación (Tabla 2).
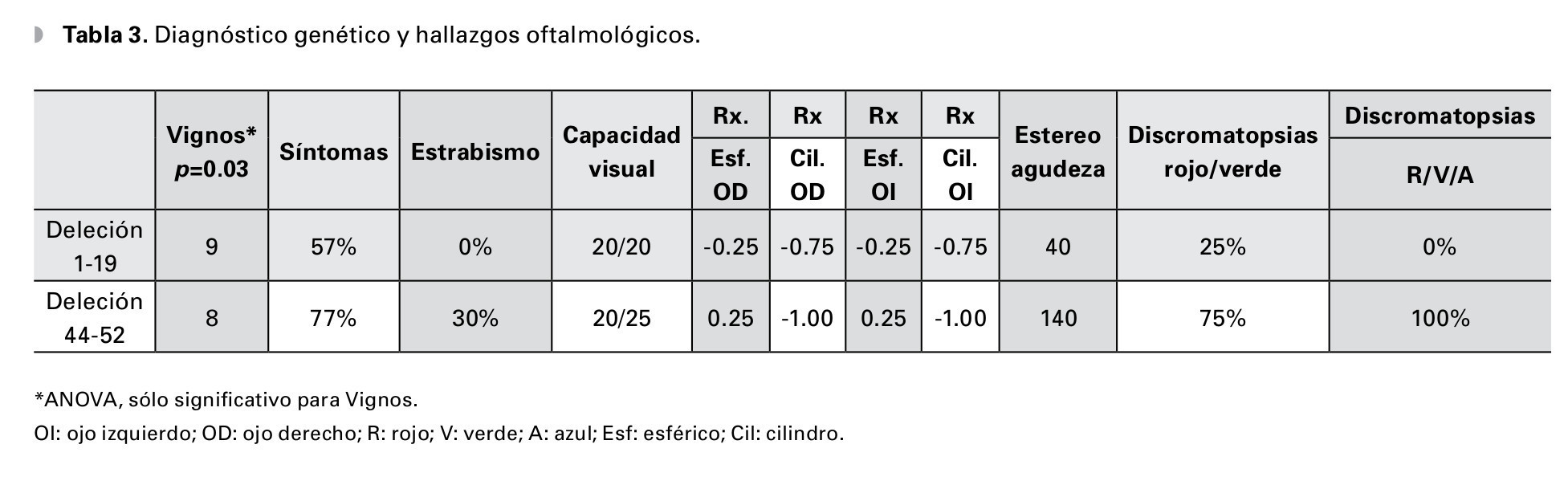
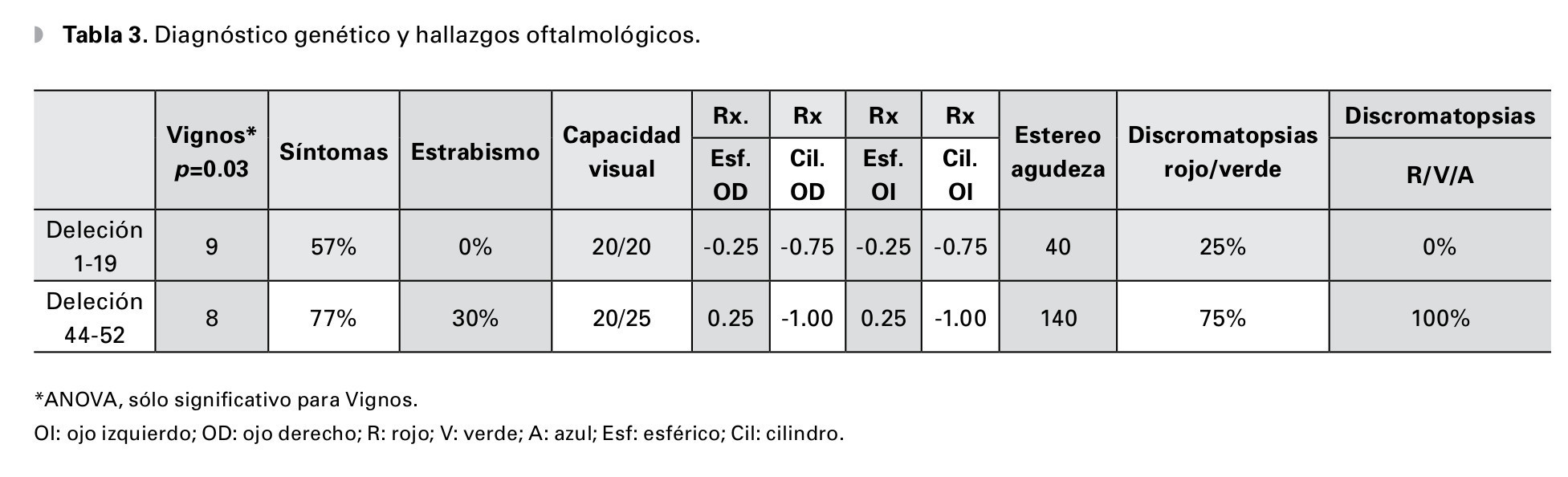
En cuanto al diagnóstico molecular de DMD hubo diferencias en severidad de las manifestaciones clínicas oftalmológicas, a pesar de que las deleciones de los exones 1 al 19 tenían un estadio más avanzado de la enfermedad, el grupo del 44 al 52 fueron 20% más sintomáticos, todos los estrabismos y trastornos de la fijación se concentraron en este último grupo, la estereopsis manifestó deterioro hasta 140 segundos de arco y los trastornos de la percepción al color fueron mayores en relación al primer grupo. La capacidad visual se mantuvo en 0.0 y 0.1 LogMAR, y el defecto refractivo más frecuente fue el astigmatismo miópico simple (Tabla 3).
• Discusión
La alta incidencia de DMD ha colocado a esta enfermedad en una de las más estudiadas por la biología molecular, es del interés de muchas áreas médicas y de investigación por la amplia distribución de la distrofina como proteína estructural en el cuerpo humano, cuya ausencia conduce a daño muscular y muerte celular, su localización en el sistema nervioso central lleva a las alteraciones oftalmológicas, motivo de este estudio. En los reportes de Sitnik R. y Campiotto, se menciona el 70% de deleción como anomalía genética más frecuente, encontradas en los exones 44 al 52 en el 54%,19 lo cual coincide con nuestro grupo de pacientes, aunque con un 93% de deleciones y el 73% encontradas en exones 44 al 52. Los trastornos de los movimientos oculares ya han sido descritos por Lui Fausta y colaboradores, al realizar EOG para estudio de los movimientos sacádicos y aunque no se menciona tener relación con la fijación, nosotros podemos proponer que la dificultad para la fijación encontrada en el 19.3% de nuestros pacientes, es debida a daño del sistema de micromovimientos y microsacadas necesarios para mantener adecuadamente la mirada en el objeto de atención, pues se trata de movimientos rápidos, además están involucradas redes neuronales corticales y subcorticales a nivel de las sinapsis, donde intervienen áreas visuales occipitales V1 y V2, áreas visuales temporales y corteza parietal posterior como bases neurofisiológicas de la atención. Los estrabismos encontrados no obedecen a un patrón único, la variedad de direcciones, su ángulo pequeño y la falla en la comitancia, sugieren involucro de músculos extraoculares aislados muy probablemente debilitados.
La alteración en la transmisión a nivel de fotorreceptores se ha demostrado clínicamente con prueba de colores de Ishihara en el estudio de Fernándes Costa, mostrando defectos en el 54% de los pacientes y sobre todo en el rojo-verde; nosotros encontramos defectos en el 43% de nuestros casos en la línea rojo-verde, esto ha sido atribuido por Verriest a la degeneración de conos, sin embargo no corresponde con la conservación de la capacidad visual que observamos en nuestros pacientes, por lo que relacionándolo a los defectos corticales, ante la falla de fijación que demostramos en el presente estudio, puede también ser secundario a compromiso de las zonas extraestriadas, especialmente la V4; es evidente además la mayor susceptibilidad al defecto rojo-verde y azul, en el grupo de deleción de exones 44 al 52. La estereoagudeza en pacientes con DMD no ha sido descrita previamente, en nuestro grupo de pacientes se registró un promedio 93 segundos de arco, considerándola mediana, relacionada con el grupo de deleción en exones 44 a 52.
• Conclusiones
Los pacientes con DMD tienen manifestaciones oftalmológicas que sugieren trastornos en la percepción del color rojo-verde, deterioro del control cerebral de la fijación y la atención, involucro de los músculos extraoculares manifestado por estrabismo y secundariamente pérdida de estereopsis, los pacientes con deleción de exones 44 al 52 desarrollan las alteraciones más severas, todo relacionado con la edad y la progresión de la enfermedad. Consideramos que el compromiso de las áreas corticales visuales, participan en la diversidad de las manifestaciones oculares.
• Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
• Financiamiento
Los autores no recibieron patrocinio para llevar a cabo este artículo.
Correspondencia:
Dra. Ivonne Segura Rangel.
Copal N° 4575, Frac. Arboledas del Sur,
C.P. 44980, Guadalajara, Jal., México.
Teléfono: (33) 31342552. Fax: (33) 31342525.
Correo electrónico: isegura@teleton-occ.org.mx
