




El término “desorden en la diferenciación sexual” (DDS) representa un grupo de anormalidades en el desarrollo del tracto genitourinario, en el cual ocurre un desarrollo atípico en uno o más niveles: cromosómico, gonadal o anatómico. Los genéticamente varones 46XY pueden presentarse con genitales externos fenotípicamente femeninos o ambiguos. El síndrome de insensibilidad a los andrógenos se podría considerar una enfermedad causada por la resistencia a la acción androgénica, causada por la mutación Xq11-12, que afecta los receptores androgénicos; la presentación clínica dependerá del grado de insensibilidad, leve (masculino infértil), moderada o completa como en nuestro caso. Requiere seguimiento por psicólogo y psiquíatra para familiares y paciente, para un desarrollo psicosexual adecuado, antes y después del tratamiento quirúrgico definitivo.
El objetivo del presente artículo es realizar una revisión sistemática de los artículos publicados en la base de datos de Medline, para identificar la epidemiología e incidencia del síndrome de insensibilidad completa a los andrógenos, así como reconocer su abordaje, tratamiento y seguimiento de estos casos.
Se presenta paciente de 23 años de edad, sin antecedentes patológicos de importancia, la cual inicia su estudio a los 17 años de edad por el Servicio de Ginecología, por presentar amenorrea y falta de desarrollo en caracteres sexuales secundarios, tiene estudios de imagen sin evidenciar estructuras Müllerianas; se realiza laparoscopía diagnóstica en 2 ocasiones sin poder identificar órganos sexuales femeninos o vestigios de testículos; estudio hormonal con niveles de estrógenos y testosterona evidentemente bajos con hormona folículo estimulante (FSH), hormona luteinizante (LH) y hormona liberadora de gonadotropinas dentro de parámetros normales; el cariotipo reporta 46XY. En ese momento se ofrece apoyo psiquiátrico, se decide continuar con educación femenina, por lo que se resuelve colocación de prótesis mamaria. Finalmente, se nos interconsulta para la realización de vaginoplastía con uso de segmento intestinal como canal vaginal.
The term “disorders of sexual differentiation” (DSD) encompasses a group of abnormalities in the development of the genitourinary tract. Atypical development occurs at one or more chromosomal, gonadal, or anatomic levels. 46 XY genetic males may present with external genitals that are phenotypically female or ambiguous. Androgen insensitivity syndrome could be considered a disease caused by resistance to androgenic action due to the Xq11-12 mutation that affects the androgenic receptors. Clinical presentation depends on the degree of insensitivity: mild (infertile male), partial, or complete, as with our patient. Psychologic and psychiatric follow-up is required for both the patient and family members so there can be adequate psychosexual development before and after definitive surgical treatment.
The aim of this article was to conduct a systematic review of published reports in the MEDLINE database to identify the epidemiology and incidence of complete androgen insensitivity syndrome and to examine the approach, treatment, and follow-up of these cases.
We present herein a 23-year-old patient, with an unremarkable pathologic history, who began to be studied by the Gynecology Service at 17 years of age due to amenorrhea and lack of secondary sexual development. Imaging studies failed to show Müllerian structures. Diagnostic laparoscopy was performed on 2 occasions in which female sexual organs or vestiges of testes were unable to be identified. Hormonal study revealed obviously low levels of estrogens and testosterone, and follicle-stimulating hormone (FSH), luteinizing hormone (LH), and gonadotropinreleasing hormone were within normal parameters; 46XY karyotype was reported. Psychiatric support was then offered. It was decided that the patient would continue to be raised and treated as a female and therefore she was given breast implants. Our service was subsequently consulted for performing vaginoplasty using an intestinal segment as the vaginal canal.
El término “desorden en la diferenciación sexual” (DDS), recientemente acuñado para lo que antes se conocía como: “estado intersexo, hermafroditismo y seudohermafroditismo”, ha sido adoptado en la actualidad ya que los expertos a nivel mundial lo reconocen mejor por esta nomenclatura1,2; este representa un conjunto de anormalidades en el desarrollo del tracto genitourinario y se refiere a un grupo de condiciones congénitas, en el que ocurre un desarrollo atípico en uno o más niveles (cromosómico, gonadal, anatómico). Los genéticamente hombres con DDS (46,XY) pueden presentarse con genitales externos fenotípicamente femeninos, ambiguos o masculinos, como en el caso de micropene (longitud < 2.5 veces menor para su edad cronológica)3, pueden ser causados por aberraciones cromosómicas y endocrinas, reflejándose en el fenotipo sexual de un individuo. La incidencia de los DDS puede variar según el grupo étnico, por ejemplo uno de cada 5,000 recién nacidos vivos en Alemania vs. uno de cada 3,000 en Egipto; esto debido a la mayor tasa de consanguinidad4. Dentro de este grupo, la hiperplasia suprarrenal congénita y la disgenesia gonadal mixta cuentan con el 50% del total de las causas con una incidencia de 1:15,000 y 1:10,000, respectivamente, aunque puede variar considerablemente entre diferentes poblaciones5.
Presentación del casoPaciente de 23 años de edad, soltera, sin antecedentes heredo- familiares o personales patológicos de importancia para el caso; inicia su estudio a los 17 años de edad por el Servicio de Ginecología, por presentar amenorrea y falta de desarrollo en caracteres sexuales secundarios, se realizan estudios de imagen sin evidenciar estructuras Müllerianas o alteraciones renales, por lo cual ingresa a laparoscopía diagnóstica en 2 ocasiones para búsqueda de órganos sexuales internos, sin poder identificar ningún órgano sexual femenino o vestigios de testículos; se realiza estudio hormonal en ese momento mostrando niveles de estrógenos de 15 pg/dL total, los cuales se repitieron en varias ocasiones sin mostrar diferencias significativas, hormona folículo estimulante (FSH) y hormona luteinizante (LH) dentro de parámetros normales pero en niveles bajos, con testosterona siempre en niveles muy bajos (último de 1.07 ng/dL); se pidió apoyo al equipo de Genética, quienes realizan estudio de cariotipo, el cual reporta 46XY (fig. 1).

En ese momento se ofrece consejo genético, así como apoyo psiquiátrico a los familiares y paciente, quienes deciden continuar con educación femenina como lo habían hecho toda su vida ya que se reflejaba fenotípicamente como sexo femenino desde la infancia; se lleva el caso al Comité de Ética del Hospital y con el consentimiento de la paciente y familiares acuden al Servicio de Cirugía Plástica, quien realiza colocación de prótesis mamaria. Finalmente, se nos interconsulta para la realización de vaginoplastía con uso de segmento intestinal como canal vaginal.
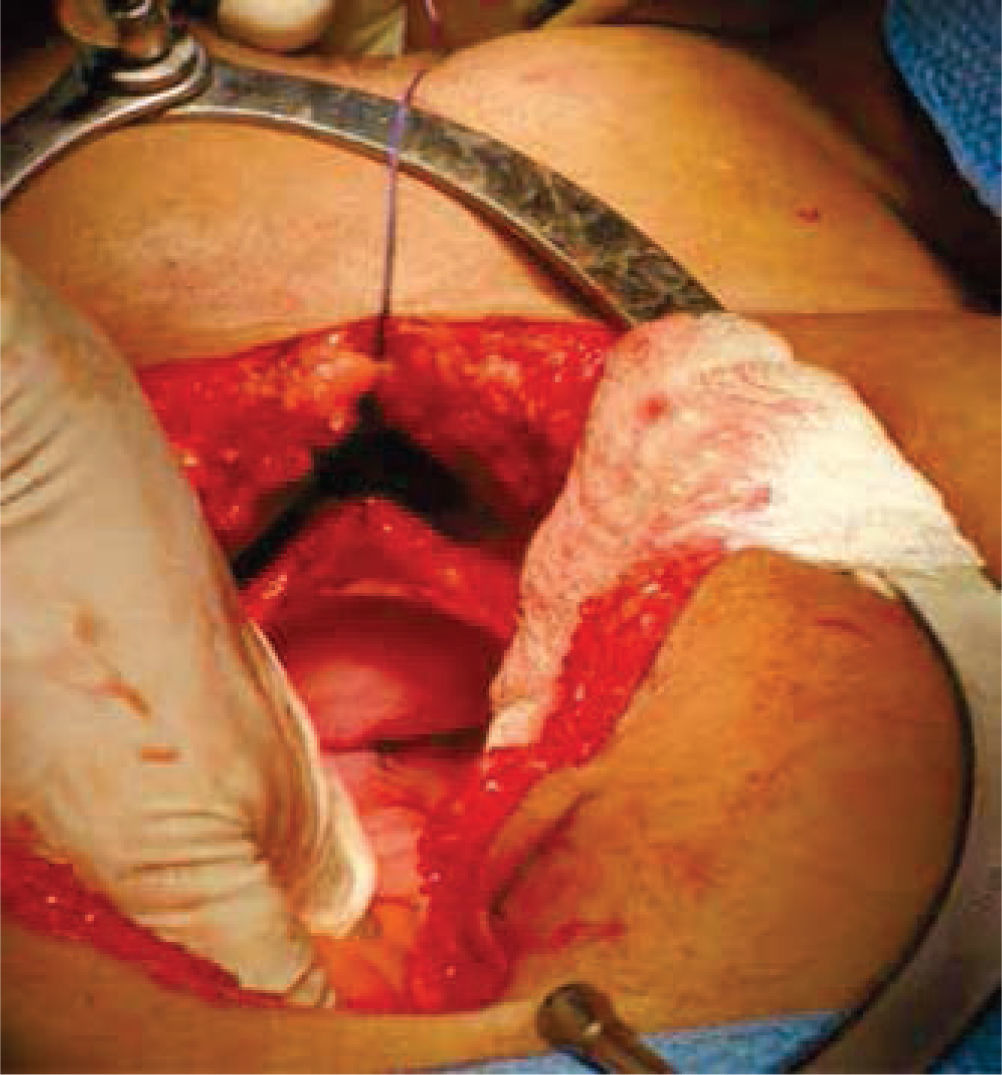
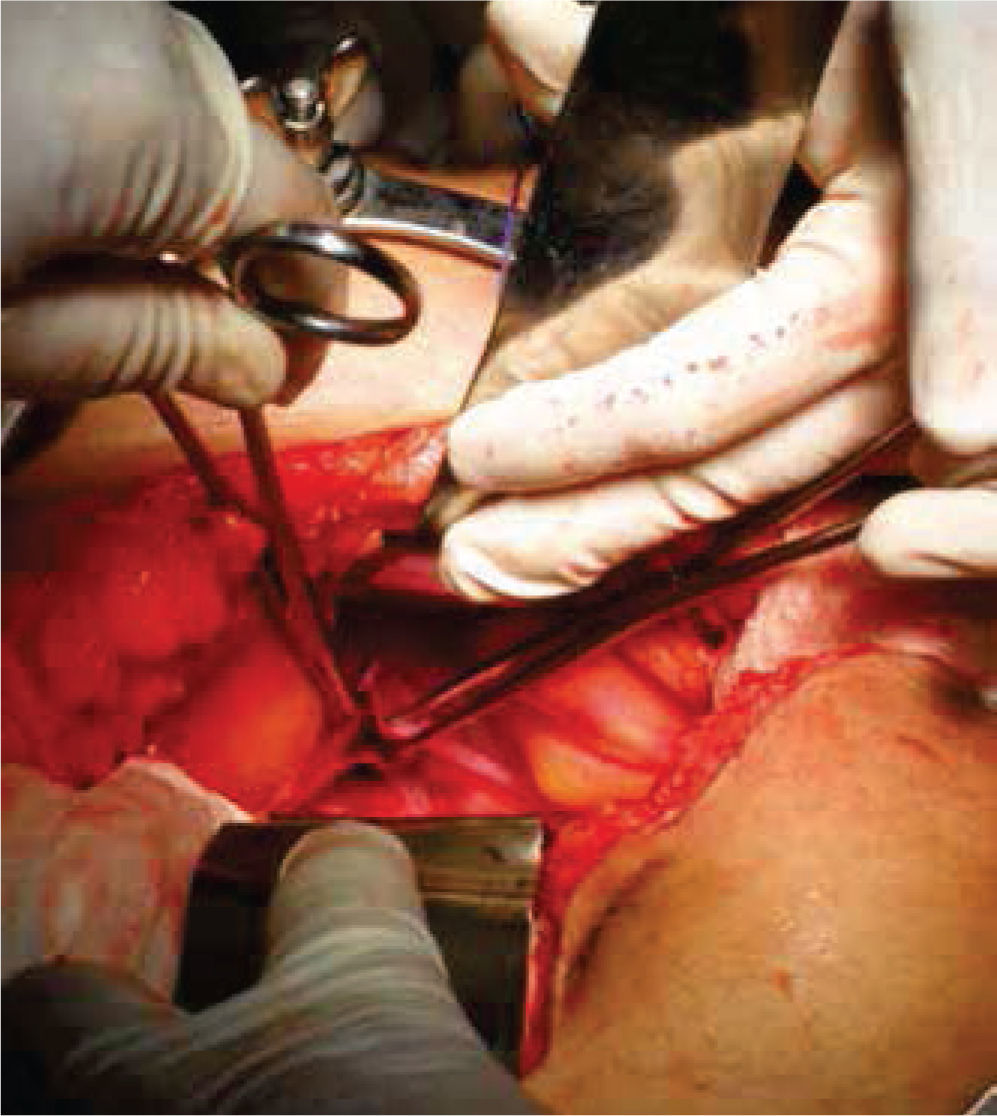
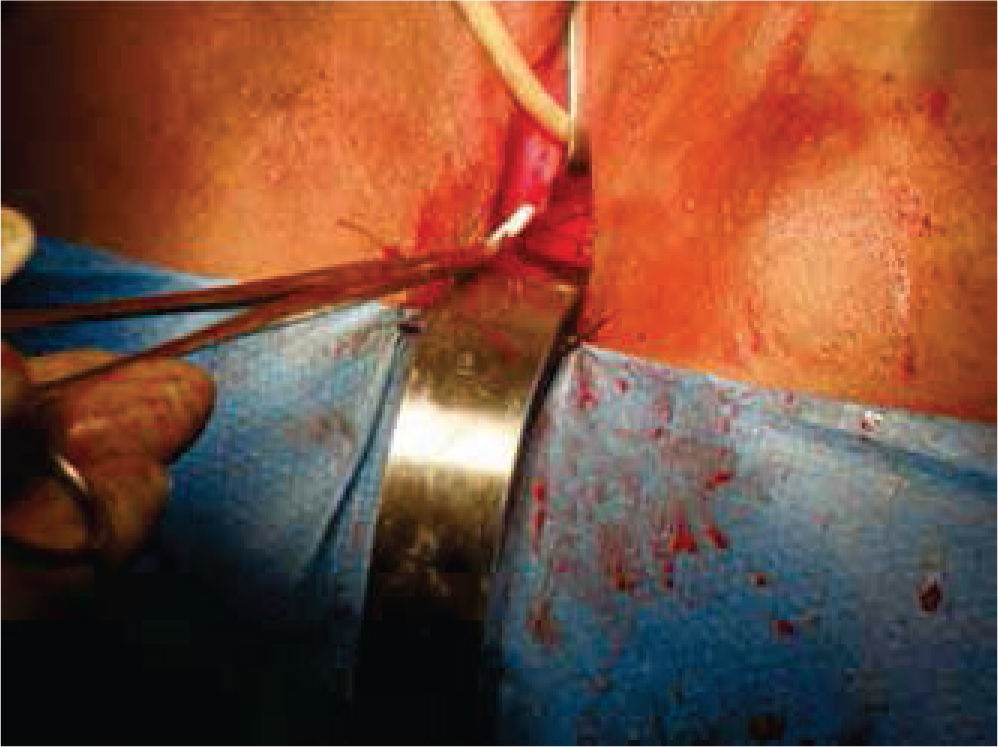
Inicialmente, se prepara a la paciente con dieta líquida un día antes de su cirugía, además de enemas evacuantes, con anestesia general y posición de litotomía, se realiza incisión media infraumbilical, disección por planos hasta llegar a cavidad abdominal, después de una minuciosa exploración se evidencia la ausencia de estructuras Müllerianas y de vestigios testiculares, con una vejiga de capacidad normal y sin próstata palpable de forma bimanual; se comienza con la apertura de la cúpula en el diafragma urogenital (figs. 2 y 3).


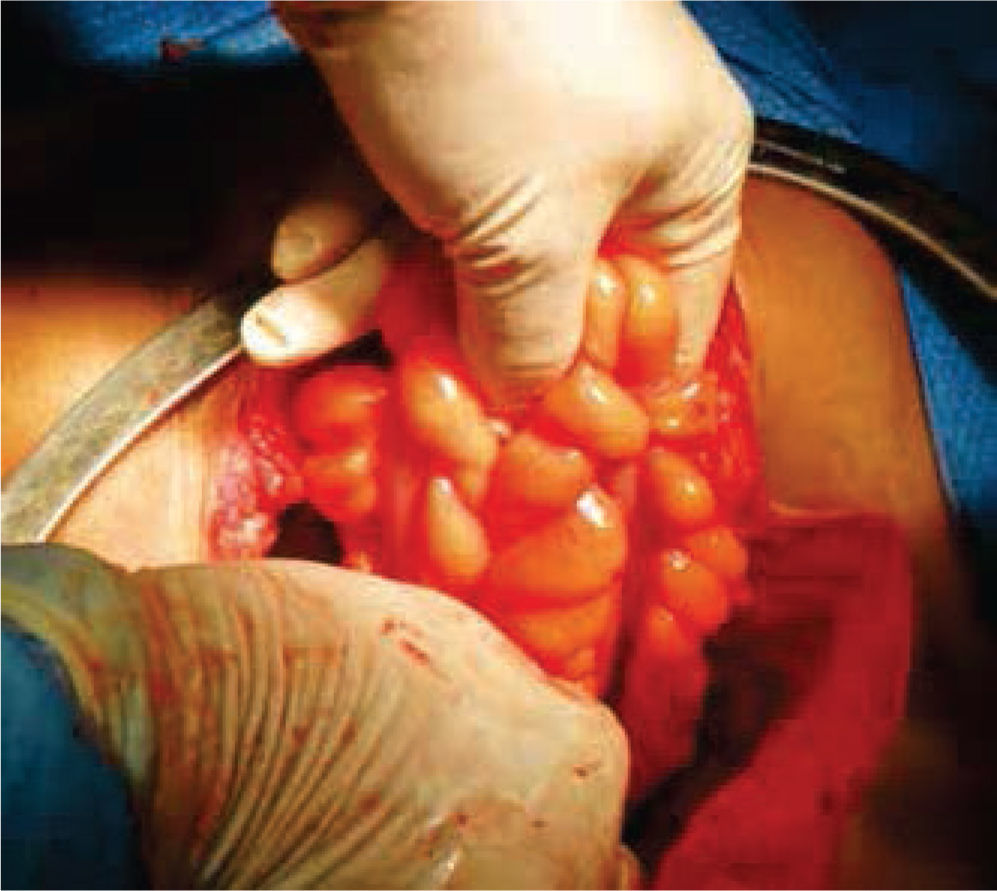
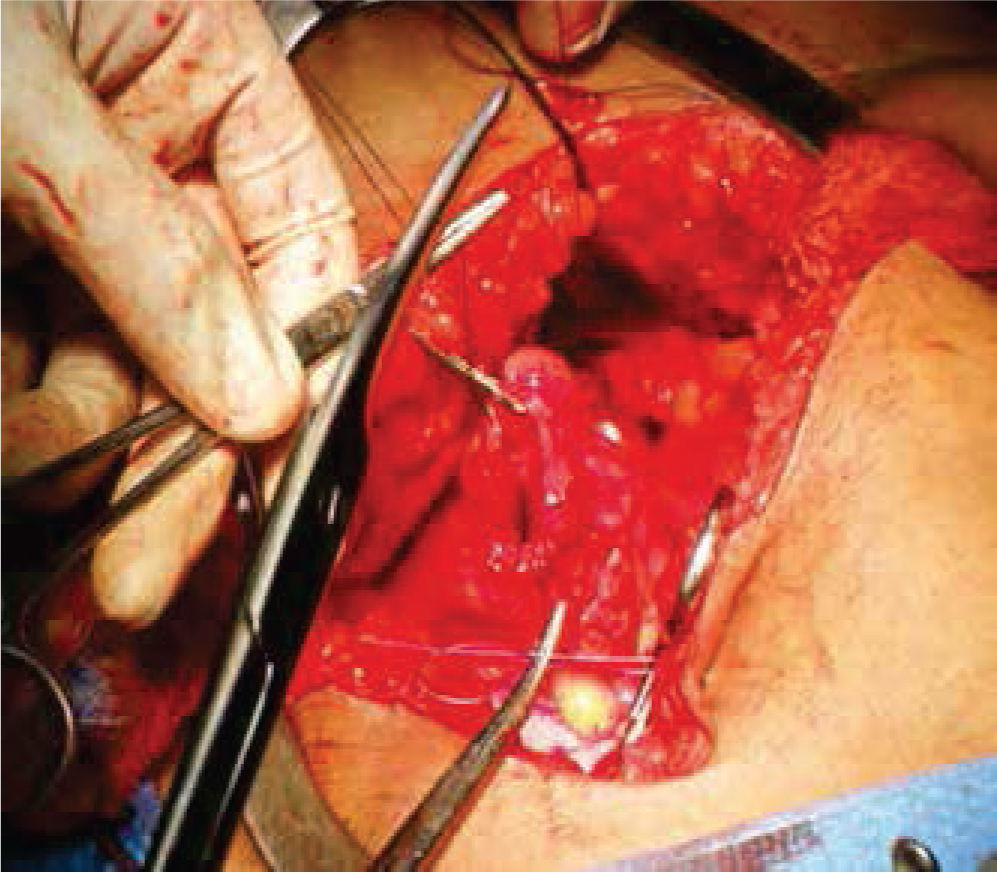
A continuación, se selecciona 12cm de sigmoides por su amplia vasculatura y mesenterio redundante, se realiza anastomosis término-terminal del asa proximal y distal al segmento resecado. Abocando el cabo proximal al neocanal vaginal, se anastomosa y se cierra el cabo distal en 2 planos (Figures. 4-6 figs.



La paciente presentó adecuada evolución por lo cual se decidió su alta en breve, en el seguimiento se indicaron dilataciones en casa con Hegar 20 minutos, 3 veces por semana, con aumento paulatino con medidas desde 8 mm hasta 22 mm al cabo de 10 meses; al seguimiento bimestral, presenta sólo leve descarga 2 veces por semana de moco, por ser segmento de colon. Continúa con seguimiento tanto psicológico como psiquiátrico, teniendo ya una vida sexual satisfactoria con su actual pareja, llevando una vida laboral normal.
DiscusiónEl síndrome de insensibilidad a los andrógenos se podría considerar una enfermedad causada por la resistencia a la acción androgénica, influenciada por 2 cosas: la morfogénesis y la diferenciación de los sistemas en el cuerpo, en los cuales tendrá la función estas hormonas. Depende en una mutación ligada al X en el gen de los receptores androgénicos, en el que puede expresar una variedad de fenotipos que va desde hombres infértiles, hasta genitales externos normales femeninos6. El primero en describir este síndrome fue John Morris7, pero no es hasta 1989 que se descubre la ubicación exacta del gen de los receptores androgénicos en Xq11-12, donde se prueba que puede presentar mutación y desarrollar la enfermedad8. Los receptores androgénicos se expresan desde la semana 8 de gestación, en la semana 9 los testes del embrión varón comienzan a secretar testosterona, teniendo 2 picos a las 11 y 18 semanas, simultáneamente se realiza la diferenciación de los epidídimos, vas deferens y vesículas seminales a partir de los conductos Wolffianos. Un andrógeno más potente, la dihidrotestosterona, se origina a partir de la testosterona por la acción de la enzima 5-alfa-reductasa tipo 2, y estimula la diferenciación del primordio genital masculino9.
Los fenotipos clínicos del síndrome de insensibilidad de los andrógenos pueden variar dependiendo de la severidad de la resistencia y se clasifican en 3 grados: completa, parcial y leve10. En particular, la resistencia completa se caracteriza por una vagina corta terminada en saco ciego, ausencia de los productos Wolffianos como epidídimo, vas deferens, vesículas seminales o próstata. La presentación clínica desde nacimiento es fenotipo femenino totalmente, haciendo difícil su diagnóstico desde un inicio11. Una pauta importante que ayuda a establecer la sospecha clínica es que, en la pubertad se presenta un crecimiento mamario generalmente pequeño o lento para la edad cronológica, con leve o ausencia de vello axilar y púbico12; otras características clínicas que se pueden encontrar son hernia inguinal mono o bilateral, aunque son mínimas las diferencias con pacientes aparentemente femeninas, lo que hace un diagnóstico difícil de sospechar desde un inicio. Este síndrome es causa del 10% del total de todas las amenorreas primarias5.
En cuanto a la presentación endocrina de estos pacientes podemos encontrar: LH normal o aumentada, y testosterona levemente por arriba de lo normal en el primer mes de vida. Después de este, la LH y la testosterona se encontrarán en niveles normales hasta la pubertad13, debido a la insensibilidad de los andrógenos y la falta de feed-back negativo de las hormonas sexuales en el hipotálamo e hipófisis. La testosterona al no tener acción se aromatiza y se convierta en estrógenos por acción enzimática más adelante, por esta razón los pacientes con insensibilidad completa cuentan con niveles más altos de estrógenos que los varones normales y desarrollan crecimiento mamario, además de poder hallarse niveles normales de hormona anti-Mülleriana, por lo cual presentan falta de caracteres sexuales femeninos internos9.
Diagnósticos diferenciales en pacientes 46XYEntre los otros diagnósticos que deben contemplarse en el caso de evaluar un individuo con DDS se encuentran: síndrome de Swyer, en el cual la falta de desarrollo de los testículos en etapas tempranas en el periodo embrionario, conlleva a la formación de caracteres sexuales femeninos como útero y trompas de Falopio por la participación de los conductos paramesonéfricos, por la falta de la testosterona y hormona anti-Mülleriana que envía el mismo testículo, quedando entonces en desarrollo sexual femenino rudimentario sin la presencia de ovarios. Entre otros síndromes diferenciales se encuentra: la feminización testicular, caracterizada por la presencia de caracteres sexuales femeninos bien diferenciados, con vagina corta, no desarrollada, y criptorquidia bilateral pero sin útero14,15. Además del ovoteste, conocido también como “hermafrodita verdadero”, el cual presenta ovarios con túbulos seminíferos16, tal como se enlistan en la tabla 1.
Diagnóstico diferencial en los trastornos de desorden en diferenciación sexual.
| Diagnósticos diferenciales | Características clínicas |
|---|---|
| Síndrome de Swyer | Presencia de útero y trompas rudimentarias |
| Feminización testicular | Caracteres sexuales secundarios femeninos, pero criptorquidia bilateral |
| Ovotestis | Presencia de túbulos seminíferos en ovarios |
Actualmente se recomienda sensibilizar a los familiares del paciente haciendo énfasis en los riesgos, beneficios y desenlaces potenciales del manejo quirúrgico, así ellos participarán de forma más activa para el cuidado de su hijo. El manejo psicosocial es parte fundamental para promover una adaptación positiva; los individuos requieren seguimiento profesional que les ayude a manejar la disfunción sexual y la dismorfia de género conforme se vaya presentando. Así como iniciar terapia de grupo, puesto que es necesario el apoyo hasta la edad adulta para poder desarrollar una vida psicosexual saludable5.
El riesgo de adquisición de enfermedad maligna en este tipo de pacientes es especialmente presentar tumores de células germinales, como gonadoblastoma, disgerminoma o seminoma, ya que todos pertenecen al mismo tipo de cáncer16, los cuales son precedidos de tejido gonadal indiferenciado17. Por lo cual, se recomienda la búsqueda intencionada y la extracción de las gónadas dismórficas a edades tempranas18.
En la actualidad, existe una base de datos internacional llamada I-DSD database, en la cual se incluyen los casos otorgados por investigadores de centros especializados, así como de reportes de casos aislados en consultorios particulares. Esta información está a disposición además de los pacientes, familiares y doctores para que sepan en dónde se encuentran los centros de investigación especializados, además de información sobre la enfermedad, seguimiento y tratamiento19.
ConclusiónEl síndrome de insensibilidad completa a los andrógenos es también parte del término de DDS, éste puede incluir pacientes desde hombres con infertilidad, hasta completo fenotipo femenino sin órganos sexuales internos como en nuestro caso.
Es una entidad rara, con impacto psicológico de los familiares así como psicosexual del paciente, por lo cual tienen que ser asesorados desde su diagnóstico para que aclaren dudas y tengan apoyo en la adaptación sexual. Requiere mantenimiento de su psicoterapeuta de por vida, además de su urólogo quien dará seguimiento de síntomas urinarios bajos y realizará exámenes de función hormonal periódica. Actualmente se encuentra una base de datos que puede ser vista por pacientes, familiares y médicos tratantes; donde encontrarán características clínicas de la enfermedad, y foros de apoyo.
Durante el tratamiento quirúrgico es importante dar asesoría amplia sobre las expectativas con el abordaje a los familiares, explicando claramente sus probables complicaciones, además de buscar intencionadamente las gónadas dismórficas hipofuncionales y extraerlas, por la probable transformación maligna ya antes referida.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
FinanciamientoNo se recibió patrocinio para llevar a cabo este artículo.