


A síndrome de neoplasia endócrina múltipla tipo 1 (MEN1) é transmitida de forma autossómica dominante e caracteriza‐se pela presença de tumores das glândulas paratiroideias, pâncreas endócrino e hipófise. Outros tumores encontram‐se menos frequentemente associados: adenomas da suprarrenal, tumores carcinoides, feocromocitomas, angiofibromas, lipomas, colagenomas e meningiomas. A presença de 2 ou mais tumores permite o diagnóstico clínico.
Apresenta‐se o caso de um homem de 56 anos, referenciado à consulta de endocrinologia para investigação de patologia nodular da tiroideia, assintomática, com aspetos benignos na ecografia e citologia aspirativa. Ao exame físico eram evidentes traços faciais grosseiros e mãos grandes. O doente tinha diagnóstico prévio de hipertensão arterial, litíase renal, síndrome de apneia obstrutiva do sono e tinha sido submetido recentemente a excisão de pólipo do cólon (benigno). A história familiar era negativa para patologia endócrina. A avaliação endócrina revelou aumento da hormona de crescimento e IGF‐1 e a prova de tolerância à glicose oral confirmou o diagnóstico de acromegalia. Os níveis séricos das restantes hormonas adeno‐hipofisárias eram normais. A ressonância magnética craniana e selar revelou macroadenoma hipofisário esquerdo e ainda uma volumosa massa extra‐axial frontal esquerda, sugestiva de meningioma. Dada a coexistência de acromegalia e meningioma, foi equacionada a hipótese de síndrome MEN1 e averiguada a presença de endocrinopatias associadas. Os níveis séricos de cálcio e paratormona encontravam‐se aumentados, com função renal e níveis de vitamina D normais, sugerindo hiperparatiroidismo primário. A insulinemia e gastrinemia eram normais. O estudo genético não identificou mutações no gene MEN1.
As manifestações fenotípicas e comorbilidades associadas sugeriram o diagnóstico de acromegalia, confirmado laboratorialmente. A associação pouco habitual de acromegalia, meningioma e hiperparatiroidismo primário sugere etiologia genética comum e permite o diagnóstico clínico de síndrome MEN1, com implicações na monitorização clínica do doente e familiares.
Multiple Endocrine Neoplasia type 1 (MEN1) syndrome is inherited as an autosomal dominant trait and is characterized by the presence of tumors in the parathyroid glands, endocrine pancreas and pituitary. Other tumors are more rarely associated: adrenal adenomas, carcinoid tumors, pheochromocytomas, angiofibromas, lipomas, collagenomas and meningiomas. The presence of two or more tumors is diagnostic of the syndrome.
We present the case of a 56 year old man referred to the Endocrine consultation for investigation of asymptomatic nodular thyroid disease, with benign features in the neck sonogram and fine needle aspiration cytology. At physical examination, coarse facial features and enlarged hands were apparent. The patient had previous diagnosis of primary hypertension, kidney stones, obstructive sleep apnea syndrome and had recent excision of a colonic polyp (benign). Family history was negative for endocrine disease. The endocrine evaluation revealed increased growth hormone and IGF‐1 and the oral glucose tolerance test confirmed the diagnosis of acromegaly. Seric levels of the remaining anterior pituitary hormones were normal. The cranial and sellar magnetic resonance revealed a left pituitary macroadenoma, and also a voluminous extra‐axial left frontal tumor, suggestive of a meningioma. Due to the coexistence of acromegaly and meningioma, the possibility of MEN1 syndrome was considered and associated endocrinopathies were investigated. Serum levels of calcium and parathormone were increased, with normal renal function and vitamin D levels, suggesting primary hyperparathyroidism. Insulinemia and gastrinemia were normal. Genetic testing did not identify mutations in MEN 1 gene.
The phenotypic manifestations of acromegaly and associated co‐morbidities were suggestive of acromegaly, which was biochemically confirmed. The unsual association of acromegaly, primary hyperparathyroidism and meningioma is suggestive of a common genetic background and is clinically diagnostic of MEN1 syndrome, which has implications in future monitoring of the patient and his relatives.
A síndrome de neoplasia endócrina múltipla tipo 1 (MEN1) é devida a mutação do gene MEN1, com transmissão autossómica dominante. Tem uma incidência de aproximadamente 0,25%. É caracterizada pela manifestação, no mesmo doente, de adenomas da paratiroideia, tumores entero‐pancreáticos e adenomas hipofisários, mais frequentemente prolactinomas. Outros tumores mais raramente associados são: adenomas do córtex suprarrenal, tumores carcinoides, feocromocitomas, angiofibromas, lipomas, colagenomas e meningiomas.
O diagnóstico desta síndrome pode ser clínico (presença de 2 ou mais tumores associados), familiar (um tumor associado à síndrome e um familiar de primeiro grau com MEN1) ou genético (portador de mutação do gene MEN1, sem manifestação clínica).
O gene MEN1, localizado no braço longo do cromossoma 11 (11q13), codifica uma proteína (menina) que regula a transcrição genética e a divisão e proliferação celulares. Estão caracterizadas 1.336 mutações, dispersas ao longo da região codificadora e locais de splicing. Em 5‐25% dos doentes com MEN1 não é possível identificar uma mutação no gene MEN1. Isto pode ser o resultado de deleção genética, ocorrência de fenocópias ou envolvimento de outros genes1.
Descrição do casoUm homem de 56 anos foi referenciado à consulta de endocrinologia por doença nodular da tiroideia. Não apresentava sintomatologia compressiva cervical ou sugestiva de disfunção tiroideia. Havia, contudo, realizado uma ecografia cervical, que mostrava glândula tiroideia de dimensões discretamente aumentadas, com nódulo único, sólido e bem delimitado, com cerca de 15mm de maior diâmetro, no lobo direito. Não eram aparentes adenomegalias a nível ecográfico. A função tiroideia era normal e o exame citopatológico do nódulo revelou benignidade.
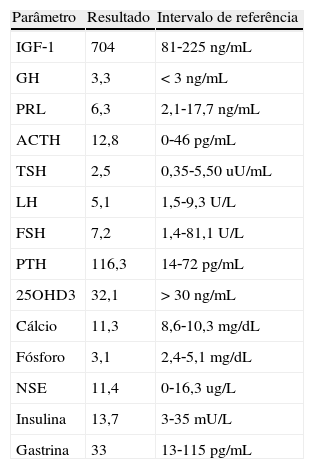
Ao exame físico, os traços faciais grosseiros, com proeminência do nariz e da mandíbula, e mãos com dedos largos levantaram a suspeita de acromegalia. A avaliação endócrina revelou hormona do crescimento (GH) e insulin‐like growth factor (IGF‐1) aumentadas (3,3ng/ml e 704ng/ml, respetivamente). A restante função hipofisária era normal, incluindo o doseamento de prolactina – tabela 1. Com a prova de tolerância à glicose oral (PTGO) com 75g de glicose não se verificou supressão dos níveis séricos de GH (persistentemente acima de 3,0ng/ml), confirmando assim o diagnóstico de acromegalia.
Resultado da avaliação laboratorial endócrina
| Parâmetro | Resultado | Intervalo de referência |
| IGF‐1 | 704 | 81‐225ng/mL |
| GH | 3,3 | <3ng/mL |
| PRL | 6,3 | 2,1‐17,7ng/mL |
| ACTH | 12,8 | 0‐46pg/mL |
| TSH | 2,5 | 0,35‐5,50uU/mL |
| LH | 5,1 | 1,5‐9,3U/L |
| FSH | 7,2 | 1,4‐81,1U/L |
| PTH | 116,3 | 14‐72pg/mL |
| 25OHD3 | 32,1 | >30ng/mL |
| Cálcio | 11,3 | 8,6‐10,3mg/dL |
| Fósforo | 3,1 | 2,4‐5,1mg/dL |
| NSE | 11,4 | 0‐16,3ug/L |
| Insulina | 13,7 | 3‐35mU/L |
| Gastrina | 33 | 13‐115pg/mL |
Dos antecedentes pessoais salientavam‐se: diagnóstico de hipertensão arterial 4 anos antes, cumprindo terapêutica diária com anti‐hipertensor em monoterapia (clortalidona 50mg/dia), com perfil tensional dentro da normalidade; litíase renal conhecida há 6 anos, com vários episódios de cólica renal e submetido a tratamento com litotrícia; excisão recente de pólipo do cólon detetado em colonoscopia de rotina, cujo resultado anátomo‐patológico revelou benignidade; síndrome de apneia obstrutiva do sono (SAOS) moderada, diagnosticada há 2 anos, sob ventilação mecânica noturna não invasiva.
Não havia história familiar conhecida de tumores hipofisários ou outra patologia endócrina.
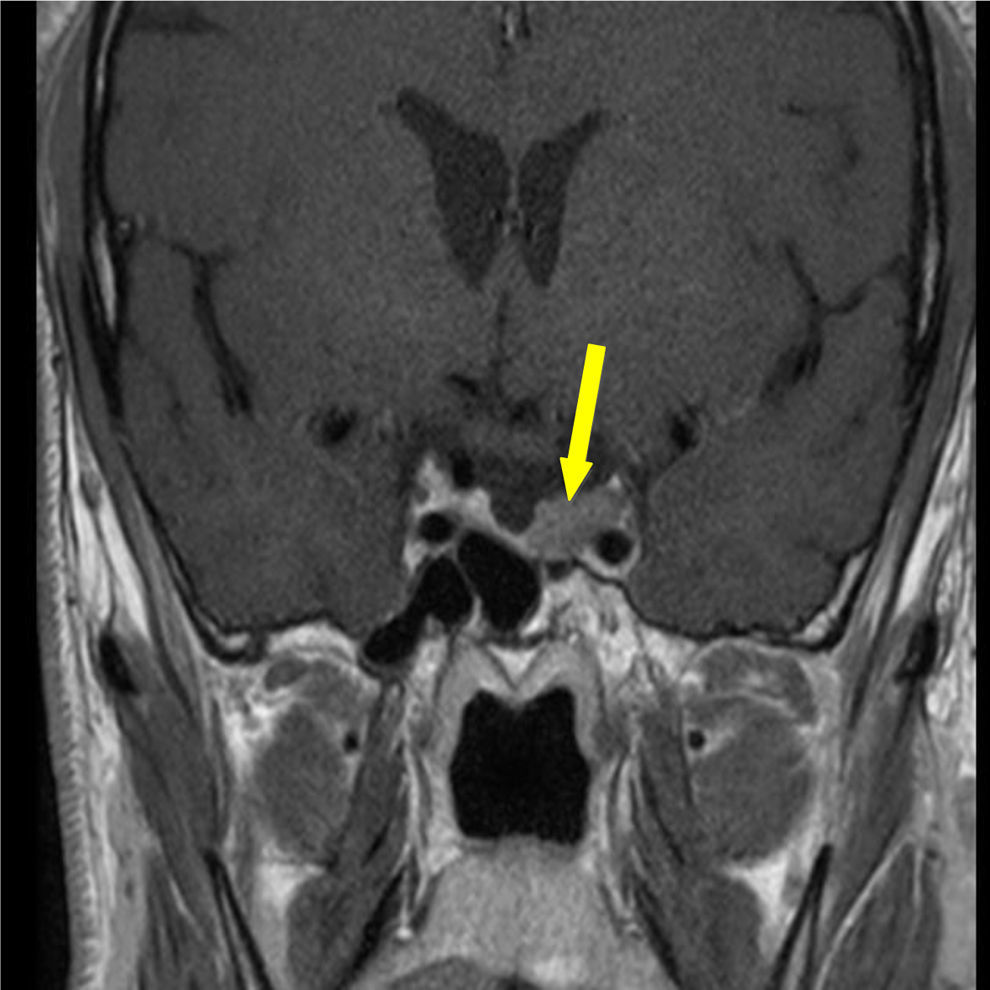
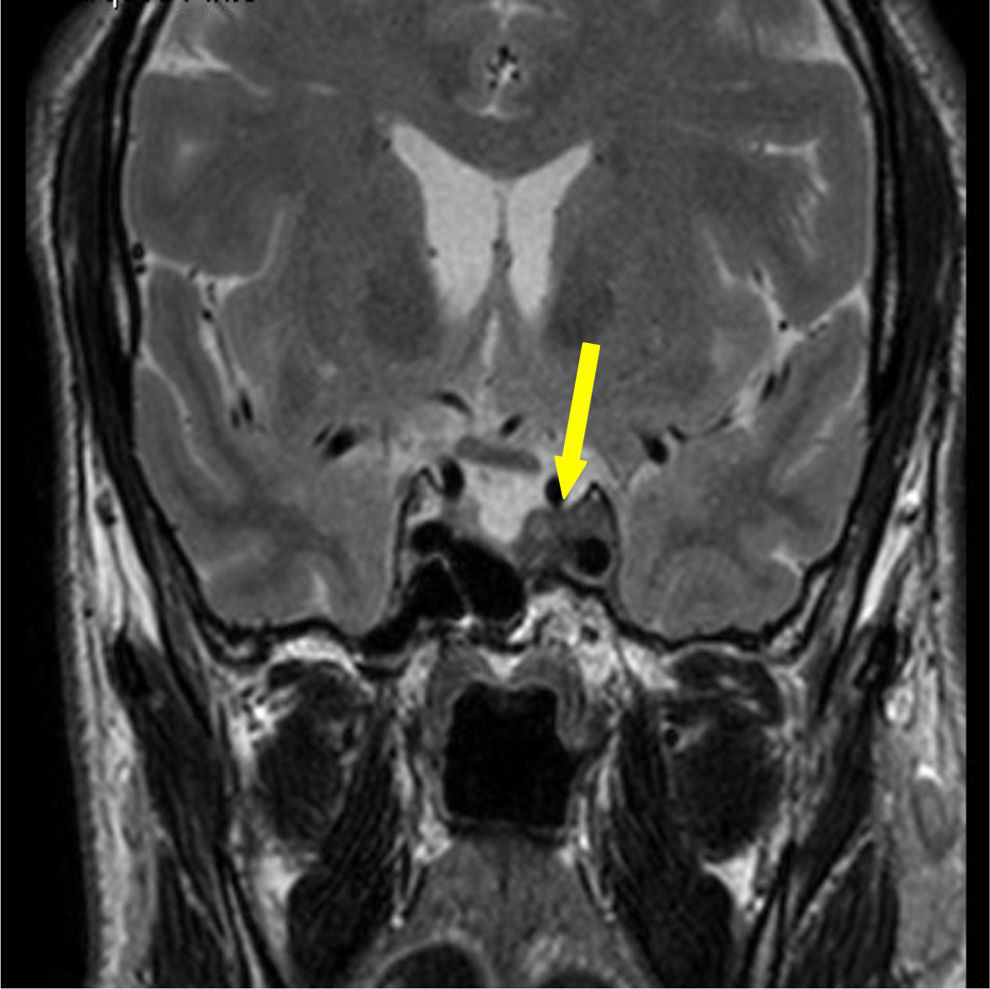
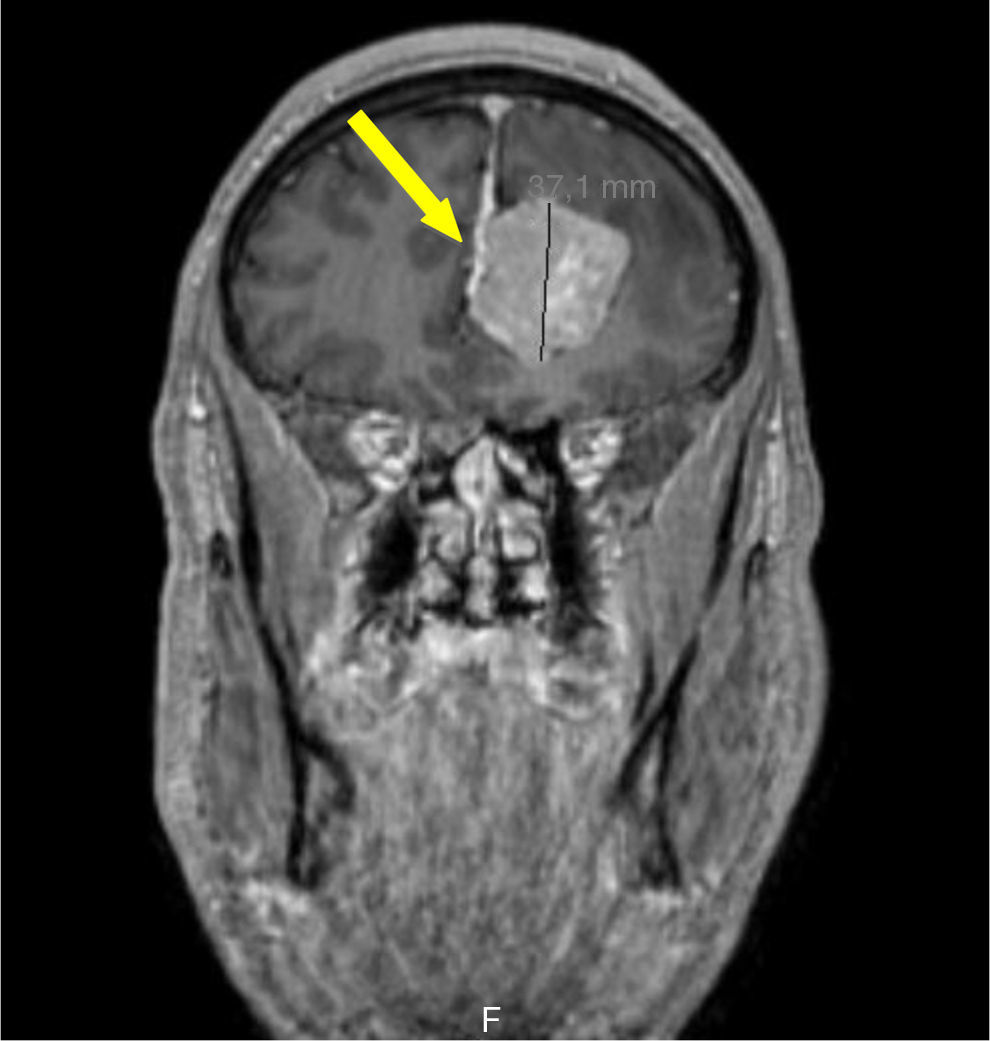
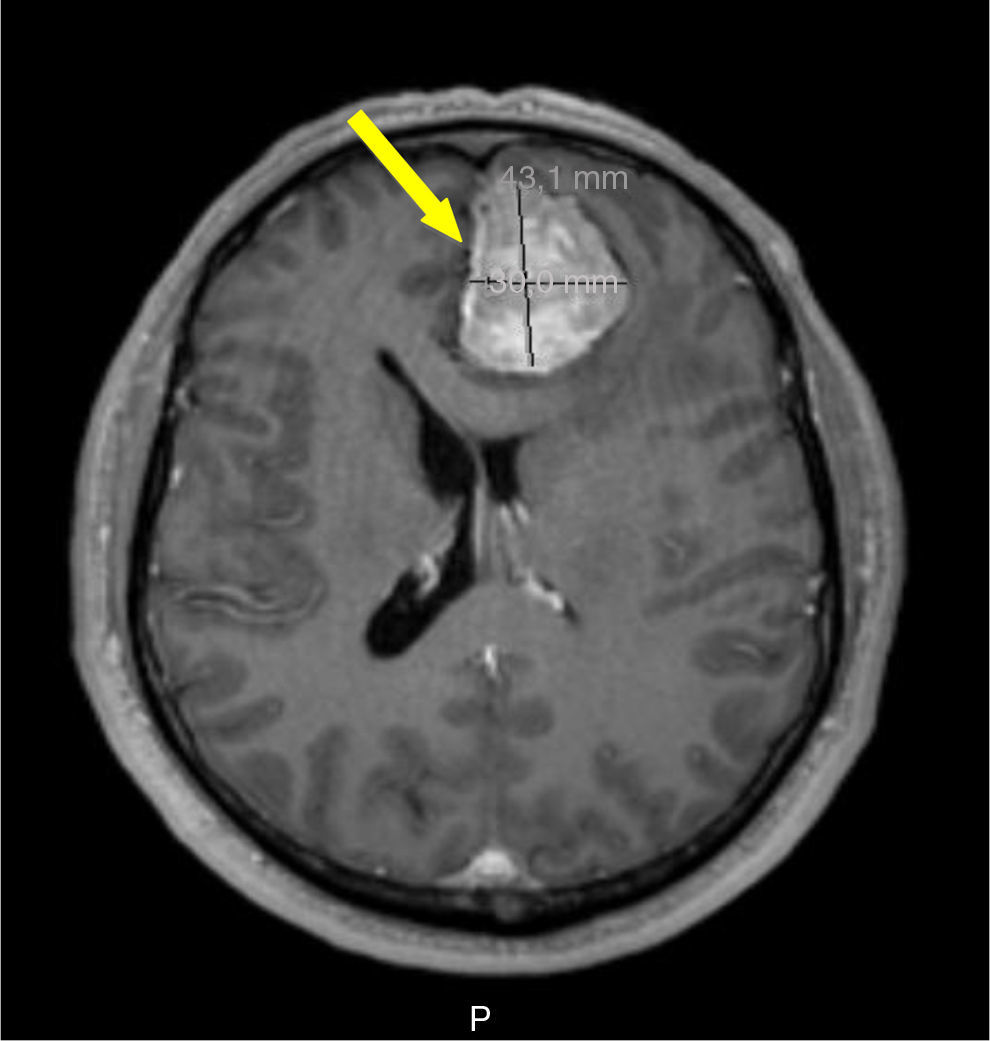
Foi realizada ressonância magnética (RMN) craniana e da sela turca que revelou uma massa selar esquerda com 15x7mm de diâmetro, induzindo ligeiro desvio da haste pituitária e com extensão ao seio cavernoso homolateral, confirmando a presença de adenoma pituitário – figs. 1 e 2. Era também evidente uma volumosa massa extra‐axial frontal esquerda, com 43x37mm, com efeito de massa e edema perilesional, sugestiva de meningioma – figs. 3 e 4.
. RMN selar, perfil coronal, ponderação T1, com gadolínio.")
. RMN selar, perfil coronal, ponderação T2.")
. RMN cerebral, perfil coronal, ponderação T1, com gadolínio.")
. RMN cerebral, perfil transversal, ponderação T1, com gadolínio.")
Perante o diagnóstico de acromegalia e meningioma foi equacionada a hipótese de síndrome MEN1. Os níveis séricos de insulina e gastrina eram normais. Os níveis séricos de cálcio e paratormona (PTH) encontravam‐se aumentados (11,3mg/dl e 116,3pg/ml, respetivamente), com nível normal de vitamina D (1,25(OH)2 D3), tendo‐se assim diagnosticado hiperparatiroidismo primário (HPTP). Em avaliações subsequentes os valores de calcemia foram de 10,6 e 10,9mg/dL. A ecografia cervical mostrou, adjacente à porção inferior do lobo esquerdo da tiroideia, uma imagem arredondada, ecogénica, sólida, com cerca de 10mm, que poderia corresponder a glândula paratiroideia aumentada. A cintigrafia com Sestamibi não revelou hiperfixação focal. O estudo genético (amplificação por polimerase chain reaction – PCR – e sequenciação da região codificadora do gene MEN1) não identificou mutações.
O doente foi avaliado em consulta de neurocirurgia e foi programada cirurgia para remoção do meningioma, que decorreu sem intercorrências e permitiu a excisão completa da lesão. O exame anátomo‐patológico confirmou o diagnóstico de meningioma meningotelial. Devido à extensão do adenoma hipofisário ao seio cavernoso, foi protelada a abordagem cirúrgica desta lesão. O doente iniciou terapêutica médica com análogo da somatostatina (Sandostatina LAR 20mg/mês) com resposta bioquímica favorável, apresentando, ao fim de 8 semanas de tratamento: IGF‐1 – 253ng/ml e GH – 0,43ng/ml. Na RMN realizada cerca de um ano após a excisão cirúrgica do meningioma e início da terapêutica médica com análogo da somatostatina não se verificava recidiva local na loca operatória e o exame era sobreponível ao anterior no que se referia a dimensão e extensão às estruturas adjacentes da lesão selar.
ComentárioNeste caso, a suspeita de acromegalia surgiu devido a traços fenotípicos sugestivos num doente avaliado por patologia aparentemente não relacionada (nódulos da tiroideia). Analisando os antecedentes pessoais do doente verifica‐se que, nos últimos 6 anos, foram diagnosticadas diversas comorbilidades frequentemente associadas àquela doença: litíase renal, hipertensão arterial, SAOS, polipose intestinal, bócio multinodular. Os doseamentos basais elevados de GH e IGF‐1 e a ausência de supressão da secreção de GH após PTGO confirmaram o diagnóstico de acromegalia. Na investigação imagiológica confirmou‐se a presença de macroadenoma hipofisário, mas o principal achado foi um volumoso meningioma com quase 5cm de maior diâmetro, que mereceu tratamento neurocirúrgico urgente. Devido à extensão do adenoma hipofisário a estruturas adjacentes do sistema nervoso central, optou‐se por iniciar tratamento médico com análogos da somatostatina para controlo bioquímico e eventual redução de tamanho da lesão, com vista a facilitar uma posterior abordagem cirúrgica. O diagnóstico de HPTP foi efetuado com base nos achados laboratoriais de elevação da calcemia e PTH, sem etiologia secundária aparente. Devido aos valores de calcemia discretamente aumentados (<1mg/dL acima do limite superior do normal), optou‐se por manter vigilância clínica e laboratorial.
A síndrome MEN1 caracteriza‐se pela ocorrência de adenomas da paratiroideia em cerca de 90% dos doentes, tumores entero‐pancreáticos em 30‐70% e adenomas pituitários em 30‐40%, dos quais o mais frequente é o prolactinoma. Outras manifestações da síndrome são a ocorrência de tumores do córtex adrenal (40%), carcinoides brônquicos, tímicos e gástricos (2‐10%), lipomas (30%), angiofibromas (85%), colagenomas (70%) e meningiomas (8%). Segundo as mais recentes recomendações para abordagem dos doentes com esta síndrome, a presença no mesmo doente de 2 ou mais tumores associados, mesmo na ausência de história familiar ou mutação no gene MEN1, permite o diagnóstico clínico1.
O estudo genético não identificou mutações da região codificadora do gene MEN1, o que pode ser atribuído à presença de mutações noutro local do gene, deleções ou presença de fenocópias. O método utilizado (PCR e sequenciação da região codificadora) apresenta algumas limitações, não identificando, por exemplo, deleções extensas do gene, que podem ser identificadas através de uma técnica complementar Multiplex Ligation‐dependent Probe Amplification (MLPA). A ausência de mutações no gene MEN1 não representa, contudo, critério de exclusão do diagnóstico, já que em 5‐25% dos casos não são detetadas mutações germinativas1. Uma outra possibilidade para a associação de adenoma hipofisário e adenoma da paratiroide no mesmo doente é a mutação no gene CDNK1B (síndrome MEN4), sendo nesse caso o codiagnóstico de meningioma um achado independente, o que nos parece menos provável.
Os meningiomas têm uma incidência anual de 2‐8:100.000, superior no sexo feminino (ratio 2:1) e na sétima década de vida. Representam cerca de 20% dos tumores do sistema nervoso central na população em geral. São na generalidade tumores benignos, de crescimento lento. Os fatores associados a maior risco de desenvolver meningioma são: traumatismo craniano, irradiação craniana e deleções no gene NF2. A maior parte tem bom prognóstico e a cirurgia e radioterapia adjuvante são curativas2.
A prevalência de meningiomas em contexto de MEN1 foi especificamente investigada e era de 8% numa população de 74 doentes3. Estudos prévios reportaram a ocorrência de meningiomas em doentes com MEN14,5, embora não tendo sido descritos como fazendo parte da síndrome. Há também relatos da associação de meningiomas com hiperparatiroidismo6,7 ou com tumores da hipófise7–12. A irradiação craniana por este ou outros motivos tem sido implicada na génese da ocorrência de meningiomas13,14, mas existem casos reportados de meningiomas e adenomas hipofisários no mesmo doente sem tratamento de radioterapia prévio, sendo menos óbvia a origem desta associação.
Desde que se verificou a associação entre acromegalia e meningiomas tem‐se vindo a especular que a GH ou outros fatores de crescimento possam ter um papel no aparecimento e crescimento dos meningiomas. Foi demonstrado in vitro que existem recetores de GH à superfície dos meningiomas e que a ativação do eixo GH/IGF‐1 aumenta significativamente o seu crescimento15. Posteriormente foi demonstrado in vivo o efeito antitumoral sobre os meningiomas, com diminuição do crescimento e por vezes regressão, após tratamento com o antagonista do recetor de GH pegvisomant16. Outros fatores estarão envolvidos neste processo, pois o aparecimento e crescimento de meningioma após tratamento médico eficaz da acromegalia foi também reportado12.
Este caso ilustra o característico elevado tempo de latência até ao diagnóstico de acromegalia, realizado no contexto de investigação clínica por outro motivo. A associação entre acromegalia e meningioma, em particular sem ocorrência de irradiação craniana prévia, é rara, com poucos casos descritos na literatura. A apresentação clínica de hiperparatiroidismo, adenoma hipofisário e um segundo tumor do sistema nervoso central não é a mais típica na síndrome MEN1, mas reúne critérios que permitem o seu diagnóstico pelas recomendações atuais, com implicações na vigilância clínica do doente e seus familiares.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
Os autores apresentam mais os sinceros agradecimentos à Sociedade Portuguesa de Endocrinologia, Diabetes e Metabolismo (SPEDM) pela atribuição de uma bolsa para deslocação ao 15.° Congresso Europeu de Endocrinologia, que se realizou em Copenhaga, de 27 de abril a 1 de maio de 2013, para apresentação deste trabalho naquele evento.
