


O atingimento do sistema nervoso central pela sarcoidose é uma entidade rara, presente em cerca de 5% dos casos. A diabetes insípida, o hipogonadismo e a hiperprolactinemia são as manifestações endócrinas mais comuns.
Apresenta‐se o caso de um doente do sexo masculino, caucasiano, 40 anos de idade, com quadro clínico com evolução de 6 meses caracterizado por sede excessiva e preferência por bebidas frias, com polidipsia (5‐6 litros de água por dia) e poliúria (5‐6 litros por dia). Como antecedentes relevantes a realçar nefrolitíase. Foi pedida uma ressonância magnética da hipófise que revelou: «ausência de hipersinal da hipófise posterior na ponderação T1 e alargamento da haste hipofisária». O doente foi então orientado à consulta de endocrinologia, tendo ficado internado para efetuar prova de restrição hídrica. Confirmou‐se diabetes insípida central com défice parcial de arginina‐vasopressina (AVP), de etiologia a esclarecer. Os outros doseamentos hormonais estavam normais. Analiticamente apresentava hipercalcemia e elevação da enzima conversora da angiotensina, pelo que no internamento se pediu uma telerradiografia de tórax que mostrou adenopatia hilar bilateral e infiltrado reticulo‐intersticial difuso. Perante a suspeita clínica de sarcoidose, foram efetuados cintigrafia com gálio67, lavado broncoalveolar e biópsia transbrônquica, com confirmação deste diagnóstico. O doente foi medicado com desmopressina intranasal (20μ/dia) e prednisolona oral (40mg/dia), atualmente com remissão dos sintomas.
Os autores pretendem realçar a diabetes insípida como manifestação inicial que levou ao diagnóstico de sarcoidose, até então desconhecida neste doente. É feita uma breve revisão da literatura no âmbito destas patologias.
Central Nervous System involvement by sarcoidosis is a rare condition, present in only about 5% of cases. Diabetes insipidus, hypogonadism and hyperprolactinemia are the most common endocrine manifestations.
A 40‐year‐old caucasian male presented with a 6‐month history of progressive thirst and preference for cold water, polydipsia (5‐6 liters/day of fluids) and polyuria (5‐6 liters/day). He had a personal history of nephrolithiasis. He was ordered a pituitary magnetic ressonance image (MRI) which shown “an enlarged infundibular stalk and absence of the posterior pituitary bright spot on T1 – weighted images”. He was then referred to Endocrinology Department, where he performed a water restriction test. Central diabetes insipidus was confirmed, with partial deficit of arginine‐vasopressine (AVP), of unknown etiology. The other hormone levels were normal. In the blood analysis it was noticed hypercalcemia and elevation of angiotensine converting enzyme (ECA), so the patient was ordered a chest x‐ray which shown bilateral enlargement of hilar lymph nodes and pulmonary infiltrates. As sarcoidosis was suspected, the patient did a Ga67 – citrate scintigraphy, a bronchoalveolar lavage and a transbronchial biopsy, and the diagnosis was confirmed. The patient was started on intranasal desmopressin (20μ/day) and prednisolone (40mg/day), with symptomatic remission.
The authors want to emphasize the diabetes insipidus as the initial manifestation which led to the diagnosis of sarcoidosis, unknown in this patient. A brief revision of the literature is made focusing on these diseases.
A diabetes insípida (DI) é uma síndrome caracterizada clinicamente por excreção de grande quantidade de urina diluída e ingestão de grandes quantidades de fluidos. Existem 2 formas principais de DI: central (défice de arginina – vasopressina [AVP]) ou nefrogénica (resistente à AVP). A DI de origem central normalmente resulta de lesões que envolvam o eixo hipotálamo – neuro‐hipofisário.
A DI no contexto de sarcoidose é invulgar, uma vez que esta doença granulomatosa só afeta o sistema nervoso central (SNC) em cerca de 5% dos casos1. A sarcoidose é uma doença sistémica granulomatosa de etiologia indeterminada. A incidência estimada é de cerca de 11 por 100.000 pessoas em caucasianos e de 36 por 100.000 na raça negra, e normalmente manifesta‐se antes dos 40 anos. Pode, no entanto, ocorrer em todas as raças e idades2. Os órgãos mais frequentemente atingidos são os pulmões e gânglios linfáticos.
Quando ocorre atingimento do SNC, este é habitualmente leptomeníngeo e vascular, e pode envolver as meninges, nervos cranianos, o hipotálamo, a haste hipofisária e a hipófise3. O hipogonadismo e a hiperprolactinemia, juntamente com a DI, são as manifestações endócrinas mais frequentes4. Relativamente aos sintomas atribuíveis à neurosarcoidose, a poliúria e polidipsia são os mais comuns, uma vez que são relatados em cerca de 33% dos doentes5. A realçar que um doente com deficiência concomitante de hormona adrenocorticotrófica (ACTH) pode ter uma DI «mascarada» por deficiência de glucocorticoides6.
A DI nefrogénica no contexto de sarcoidose é também possível, uma vez que esta doença muitas vezes cursa com hipercalcemia e nefrocalcinose, 2 etiologias possíveis de DI com resistência à ação da AVP.
Os autores apresentam o caso de um doente cujo diagnóstico de sarcoidose foi feito a partir de sintomatologia inicial compatível com DI.
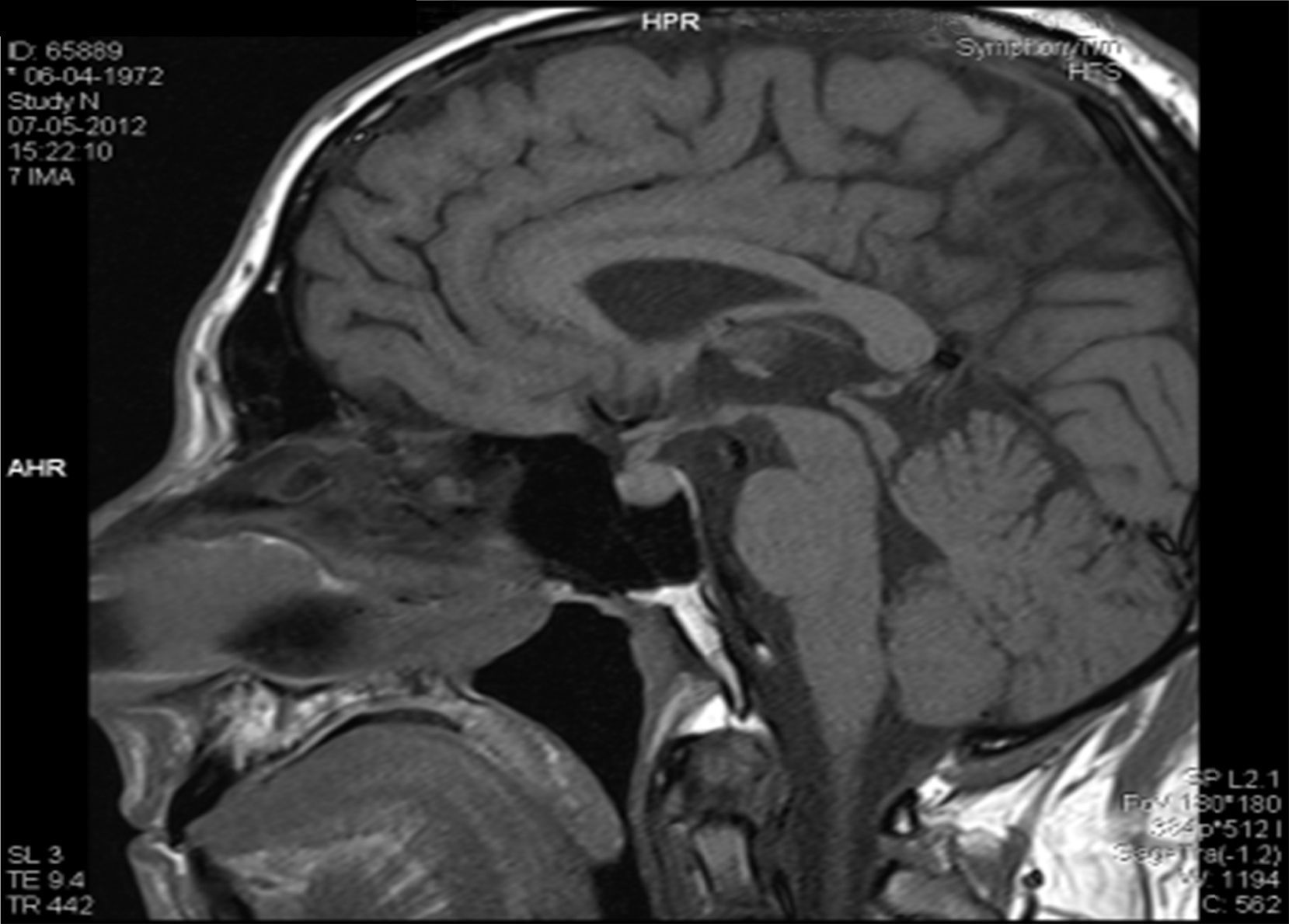
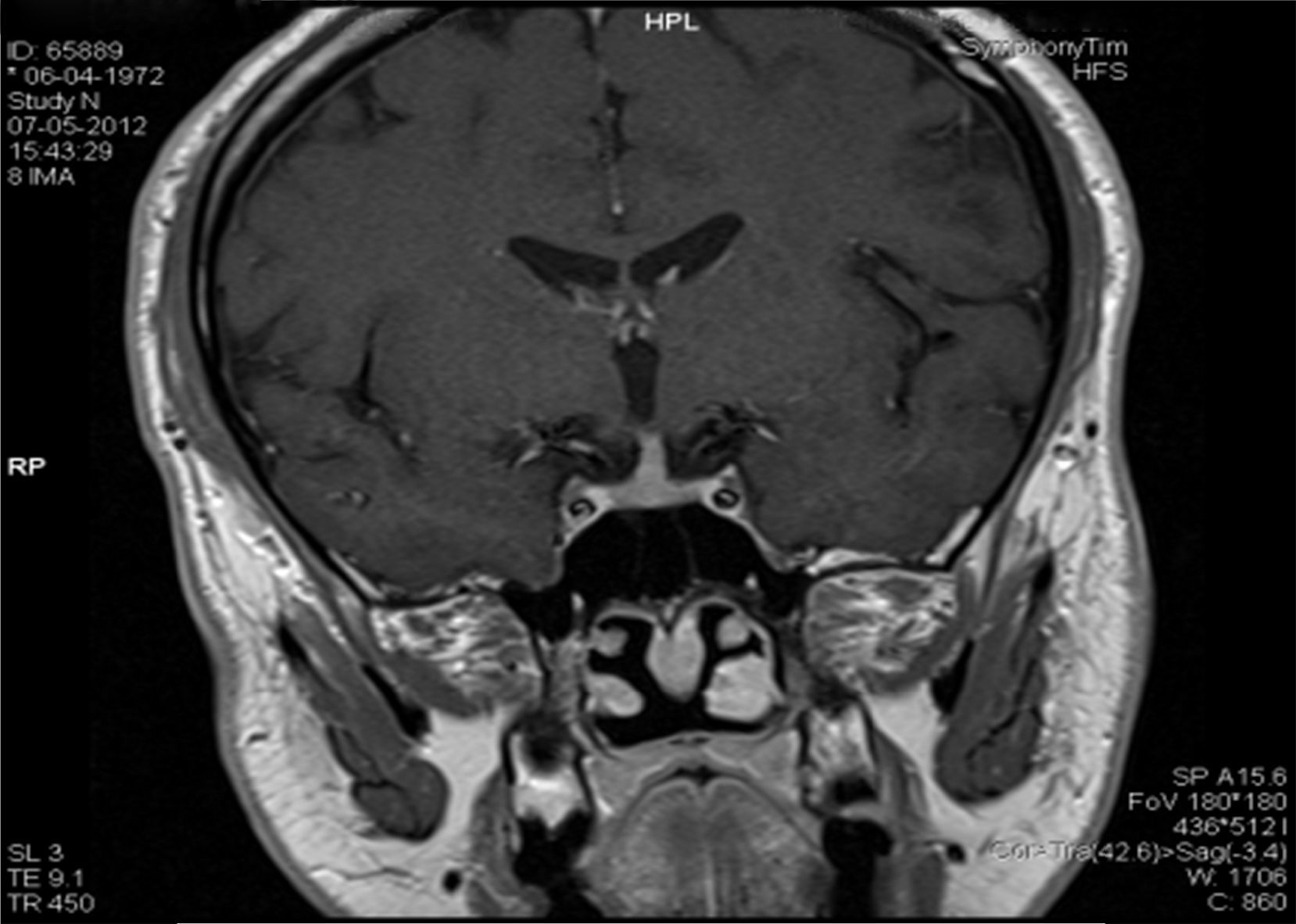
Descrição do casoDoente do sexo masculino, 40 anos, caucasiano, com antecedentes pessoais de nefrolitíase (seguido por urologia) e sem medicação habitual. Sem outros antecedentes pessoais de relevo e sem história familiar significativa. Por apresentar história de poliúria (cerca de 5‐6L por dia, levantando‐se várias vezes durante a noite para urinar) e polidipsia (cerca de 4‐5L por dia de ingestão de água, com preferência por líquidos frios), recorreu a um médico urologista, que lhe solicitou uma ressonância magnética (RM) hipofisária. Na RM constatava‐se «alargamento da haste hipofisária e ausência do habitual hipersinal da neuro‐hipófise na ponderação T1 – compatível com DI central» (figs. 1 e 2). O doente foi então referenciado à consulta de endocrinologia para estudo da sua situação clínica. Relativamente aos sintomas, o doente confirmava a poliúria e polidipsia, com evolução de cerca de 6 meses; referia também astenia e tosse seca. Negava febre e perda ponderal, assim como outra sintomatologia sistémica; negava também cefaleias, alterações visuais, desejo sexual hipoativo ou impotência. Ao exame físico não tinha alterações de relevo. Ficou internado no serviço de endocrinologia para estudo analítico e hormonal basal e realização de prova de restrição hídrica para confirmação de suspeita clínica de DI.
 em corte sagital: de notar a ausência do habitual hipersinal em T1da hipófise posterior.")
 em ponderação T1: de realçar o alargamento da haste hipofisária.")
Analiticamente foram excluídas diabetes mellitus e insuficiência renal – glicose plasmática em jejum de 85mg/dl, ureia de 40mg/dl (valores de referência [V.R.] entre 8‐50), creatinina de 0,9mg/dl (V.R.: 0,7‐1,2). O ionograma estava também normal: sódio de 140mEq/L (V.R.: 135‐145), potássio de 4,2mEq/L (V.R.: 3,5‐5). No entanto, apresentava hipercalcemia discreta, com cálcio sérico de 10,78mg/dl (V.R.: 8,9‐10,3). O estudo hormonal basal não revelava alterações: prolactina de 5μg/L (V.R.: <17μg/L), FSH de 11UI/L (V.R.: 2‐17), LH de 8UI/L (V.R.: 4‐18), testosterona total de 588ng/dl (V.R. 350‐890ng/dl), TSH de 2,2mU/L (V.R.: 0,5‐4,7), T4 livre de 0,9ng/ml (V.R.: 0,6‐1,2), ACTH às 8h da manhã de 28pg/ml (V.R.: 10‐50) e cortisol plasmático às 8h da manhã de 19,2μ/dl (V.R.: 5‐25).
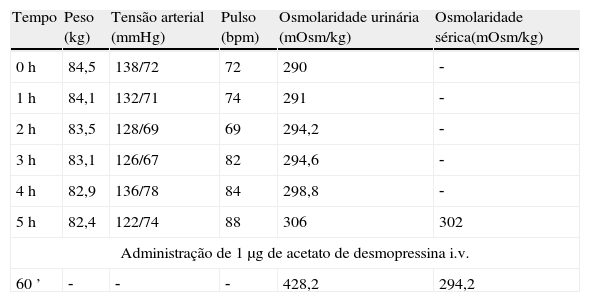
Realizou então uma prova de restrição hídrica para confirmação do diagnóstico de DI (tabela 1); a prova foi realizada no internamento, iniciou‐se de manhã cedo e o doente foi avisado para não ingerir água ou qualquer alimento. No fim de prova os valores de osmolaridade sérica e urinária foram consistentes com o diagnóstico de DI central com défice parcial de AVP, de etiologia a esclarecer.
Resultados da prova de restrição hídrica
| Tempo | Peso (kg) | Tensão arterial (mmHg) | Pulso (bpm) | Osmolaridade urinária (mOsm/kg) | Osmolaridade sérica(mOsm/kg) |
| 0h | 84,5 | 138/72 | 72 | 290 | ‐ |
| 1h | 84,1 | 132/71 | 74 | 291 | ‐ |
| 2h | 83,5 | 128/69 | 69 | 294,2 | ‐ |
| 3h | 83,1 | 126/67 | 82 | 294,6 | ‐ |
| 4h | 82,9 | 136/78 | 84 | 298,8 | ‐ |
| 5h | 82,4 | 122/74 | 88 | 306 | 302 |
| Administração de 1μg de acetato de desmopressina i.v. | |||||
| 60’ | ‐ | ‐ | ‐ | 428,2 | 294,2 |
Legenda: a prova de restrição hídrica foi suspensa quando ambos os critérios foram cumpridos – osmolaridades urinárias consecutivas com diferença inferior a 10% e perda de peso de pelo menos 2%. No fim da prova de notar que a osmolaridade sérica era inferior à urinária. Após administração de desmopressina, aumento de cerca de 40% na osmolaridade urinária.
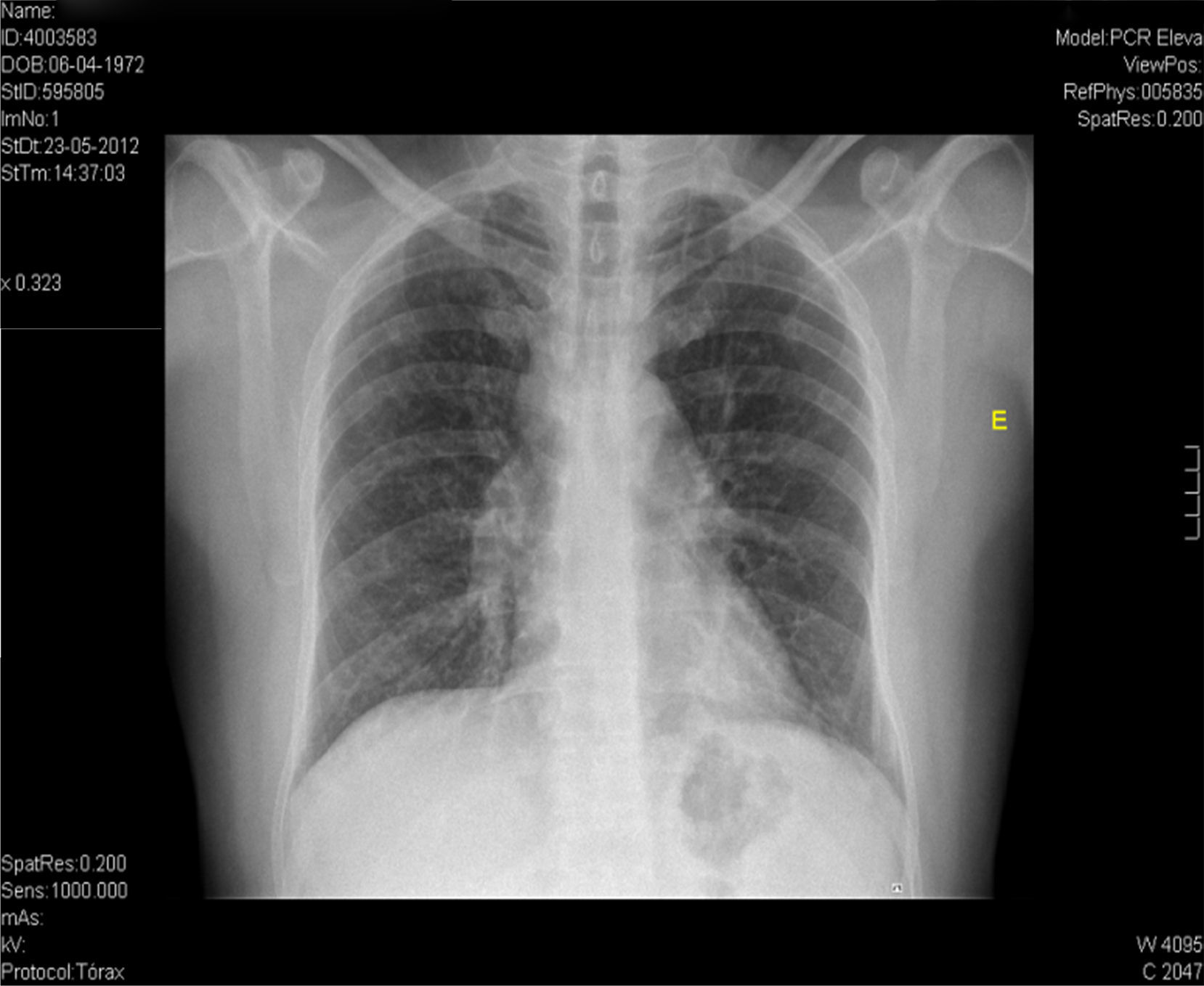
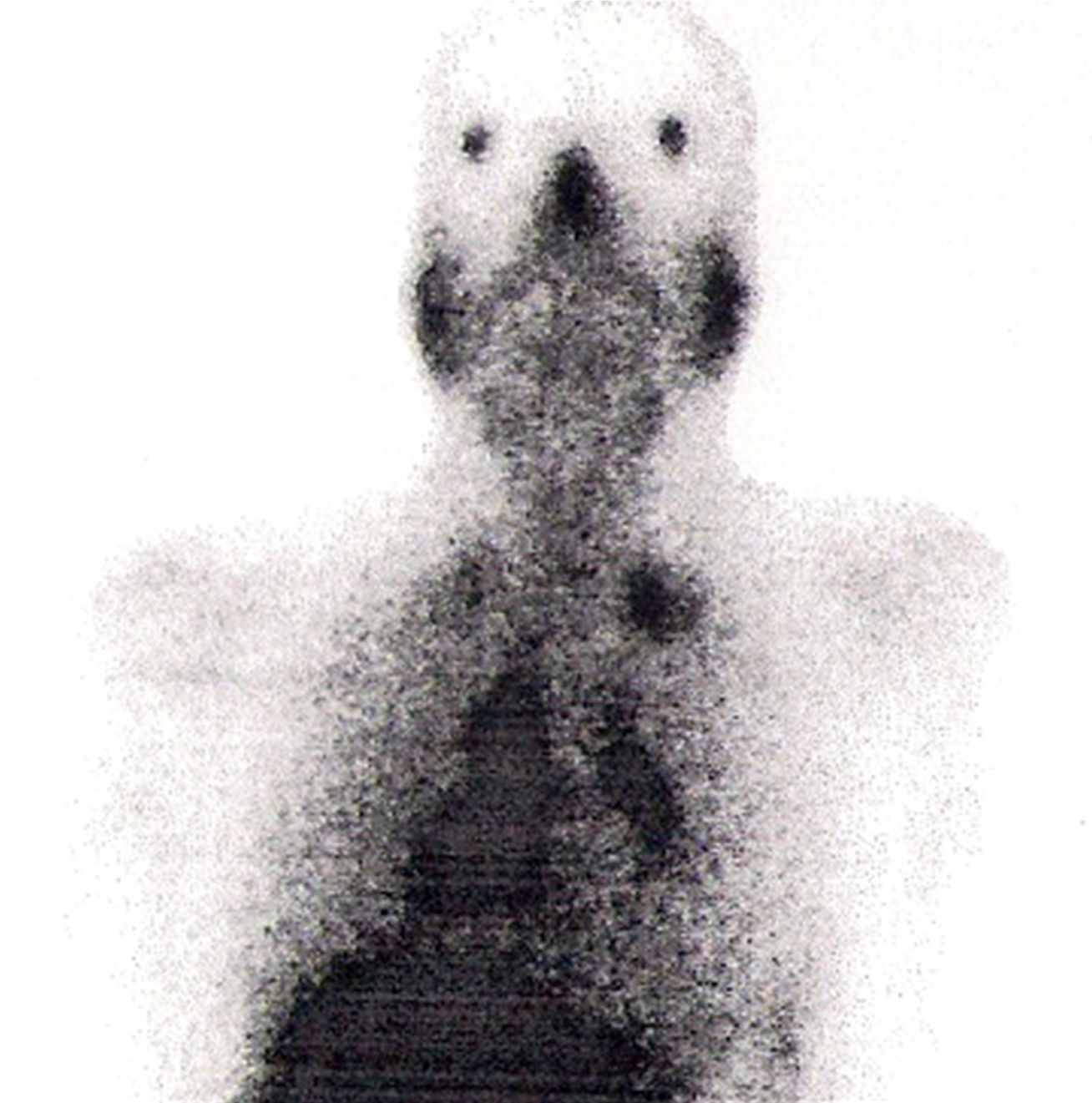
Na RM hipofisária descrita anteriormente o doente apresentava ausência de hipersinal da hipófise posterior na ponderação T1 e alargamento da haste hipofisária, o que nos alertava para uma série de diagnósticos, sendo os mais prováveis a hipofisite linfocítica ou doenças granulomatosas (sarcoidose, tuberculose, granulomatose de Wegener, histiocitose de células de Langerhans, doença de Erdheim‐Chester). Dado que o doente se queixava de astenia, tosse seca, e analiticamente apresentava hipercalcemia, foram pedidas velocidade de sedimentação (VS) e níveis séricos de enzima conversora de angiotensina (ECA). Ambos os valores se encontravam alterados, com VS de 85mm (V.R.: <15) e ECA de 319,5U/L (V.R.: 8‐52). Pediu‐se também uma telerradiografia do tórax (Rx tórax), que revelava adenopatias mediastínicas bilaterais e infiltrado reticulo‐intersticial difuso em ambos os campos pulmonares (fig. 3). Perante estes achados, a hipótese diagnóstica que se colocou em primeiro lugar foi sarcoidose com atingimento do eixo hipotálamo – hipófise. Foi efetuada uma cintigrafia com citrato de gálio67, que demonstrou uma captação do radiofármaco sugestiva de sarcoidose (sinal de Panda – fig. 4). Foi referenciado à pneumologia onde efetuou um lavado broncoalveolar, que mostrou um ratio CD4/CD8 de 8,3 (consistente com o diagnóstico) e uma biópsia transbrônquica que revelou granulomas múltiplos não caseosos e negativos para bacilos álcool‐ácido resistentes (BAAR) e para fungos.
.")
 – sugestivo de sarcoidose.")
Iniciou tratamento com 1‐desamino‐8‐D‐argina‐vasopressina (DDAVP) intranasal, na dose inicial de 10μg/dia, passando depois para 20μg/dia, com regressão dos sintomas de poliúria e polidipsia. Iniciou também terapêutica com prednisolona oral 40mg/dia, referindo melhoria franca da astenia. No seguimento deste doente será importante repetir a RM hipofisária para ver se o tratamento dirigido à sarcoidose com corticoterapia foi suficiente para reverter as alterações encontradas na neuro‐hipófise e assim dispensar a administração diária de DDAVP intranasal.
ComentáriosA DI como forma de apresentação de sarcoidose é uma entidade rara; a neurosarcoidose pode ocorrer isoladamente ou juntamente com outras manifestações sistémicas desta doença, embora cerca de 97% dos doentes com neurosarcoidose tenham doença sistémica, mesmo que esta seja desconhecida e os doentes sejam assintomáticos1.
Como já foi referido, o envolvimento do SNC na sarcoidose só acontece em cerca de 5% dos casos. A idade típica de apresentação da neurosarcoidose é entre 33‐41 anos1.
O primeiro exame complementar de diagnóstico efetuado neste doente foi a RM hipofisária, pedida pelo seu urologista face à sintomatologia característica de DI. No entanto, será mais prudente efetuar como primeiro passo na investigação a confirmação bioquímica de uma DI, uma vez que até cerca de 20% dos indivíduos saudáveis podem ter ausência de hipersinal da hipófise posterior na ponderação T17. A ausência de hipersinal em T1 normalmente corresponde a deficiência dos grânulos neurosecretores da AVP e da sua proteína transportadora, a neurofisina. A AVP é sintetizada nos núcleos paraventriculares localizados bilateralmente nas paredes do terceiro ventrículo e nos núcleos supraópticos localizados nos extremos do quiasma ótico; assim, tumores confinados à sela turca normalmente não produzem DI8. Doenças associadas a alargamento da haste hipofisária são: infundibulohipofisite linfocítica, doenças granulomatosas, metástases, craneofaringeomas e germinomas. A hipofisite linfocítica é uma entidade em crescente reconhecimento e existem formas confinadas apenas à hipófise posterior e infundíbulo (infundibuloneurohipofisite linfocítica) que podem cursar somente com DI9. As metástases (principalmente com origem nos pulmões ou mama), os craneofaringeomas e germinomas normalmente produzem massas facilmente identificadas na RM, o que não era o caso neste doente. As doenças infiltrativas e granulomatosas têm uma predileção pela hipófise posterior5; a histiocitose de células de Langerhans envolve frequentemente a neuro‐hipófise e o infundíbulo, e pode causar DI pela destruição do lobo posterior e disfunção da haste hipofisária. As doenças granulomatosas, como a tuberculose, a sarcoidose e a sífilis podem também infiltrar a hipófise posterior e causar DI10. Sendo verdade que um diagnóstico definitivo das lesões da neuro‐hipófise só é possível através de confirmação histológica, este é um processo muito invasivo e com vários riscos associados; assim, um diagnóstico presuntivo pela clínica, achados laboratoriais, imagiológicos e pela histologia das lesões pulmonares revelou‐se gratificante no sentido de eliminar a necessidade de biópsia hipofisária.
Do ponto de vista endocrinológico, a única consequência da disfunção da neuro‐hipófise é a DI central, que é uma doença benigna se o doente tiver a capacidade de repor as perdas urinárias com a ingestão equivalente de fluidos, ou seja, se o mecanismo da sede estiver intacto. O diagnóstico de DI é confirmado formalmente através da prova da restrição hídrica. Neste doente foi medida a osmolaridade urinária inicial, que era de 290mOsm/kg. O peso inicial do doente era de 84,5g Foi então efetuada Colheita horária de urina, e quando a variação entre osmolaridades urinárias consecutivas foi inferior a 10% (ou a 30mOsm) e a perda de peso foi cerca de 2% do peso inicial suspendeu‐se a prova, o que ocorreu passadas 5h (osmolaridade urinária final de 306mOsm). De realçar que as 2 primeiras urinas consecutivas já cumpriam os critérios de diferença de osmolaridades inferior a 10%, no entanto, o doente ainda não tinha atingido os critérios da perda de peso, pelo que se continuou a prova. A osmolaridade plasmática após a prova de desidratação era de 302mOsm/kg e o peso do doente era de 82,4kg Assim, administrou‐se 1μg de acetato de desmopressina intravenosa e ao fim de 60 minutos os valores eram os seguintes: osmolaridade urinária de 428,2mOsm/kg (aumento de cerca de 40%) e osmolaridade plasmática de 294,2mOsm/kg. Em doentes com formas graves de DI a osmolaridade urinária inicial é normalmente já inferior a 200mOsm/kg, o que não era o caso deste doente. Uma vez que após privação de água o doente não teve capacidade para concentrar adequadamente a urina (a diferença entre osmolaridades urinárias inicial e final foi inferior a 10%) confirma‐se o diagnóstico de DI. Uma resposta normal consiste numa osmolaridade urinária máxima entre 800‐1.400mOsm/kg no final da prova de restrição hídrica. Tanto na DI central com défice severo de AVP como na DI nefrogénica a osmolaridade plasmática no fim da prova de desidratação (antes da administração da desmopressina) é superior à osmolaridade urinária; para a diferenciação entre ambas administra‐se desmopressina ou vasopressina intravenosas. Em indivíduos saudáveis existe só um pequeno aumento (<9%) da osmolaridade urinária em resposta a este fármaco. Na DI central total existe um aumento de osmolaridade urinária superior a 50%, enquanto que na DI nefrogénica esse aumento é inferior a 45%11–12. O doseamento de níveis séricos de AVP é também útil neste contexto, uma vez que na DI central esta hormona normalmente não é detetada, enquanto na DI nefrogénica atinge valores elevados, frequentemente superiores a 5pg/ml.
Neste doente a osmolaridade plasmática antes da administração da desmopressina foi inferior à osmolaridade urinária e ao fim de uma hora após a administração de desmopressina intravenosa houve um aumento de cerca de 40% na sua osmolaridade urinária – compatível com DI central parcial. O desafio diagnóstico consiste em diferenciar DI central parcial de polidipsia primária. Em ambas as patologias existe alguma capacidade de concentrar urina com a restrição hídrica, mas a concentração urinária não se aproxima dos 800mOsm/Kg, o que é característico em indivíduos normais. Em resposta à desmopressina, indivíduos com DI central parcial têm um aumento superior a 9% na concentração urinária, enquanto indivíduos com polidipsia primária não têm aumento na concentração urinária13.
Assim, o diagnóstico mais provável neste doente será DI central parcial secundária a infiltração sarcoidótica hipotálamo‐hipofisária. Para além da confirmação bioquímica, o doente tinha também imagiologia compatível. A não esquecer que na sarcoidose pode haver também DI nefrogénica como consequência de hipercalcemia e/ou nefrocalcinose, pelo que não se pode excluir um componente de resistência renal à ação da AVP neste doente.
O objetivo principal do tratamento num doente com DI central e mecanismo da sede preservado é diminuir a polidipsia e poliúria para um nível que permita manter um estilo de vida normal. A primeira linha no tratamento farmacológico é a desmopressina, um análogo sintético da AVP (mas muito mais potente que a AVP para inibição da diurese14,15). A desmopressina pode ser administrada oralmente ou por via intranasal; a dose máxima necessária raramente é superior a 0,2μg oralmente ou 20μg por via intranasal16. A hiponatrémia é uma complicação rara. Relativamente ao tratamento da doença de base com corticóides, é controverso se pode reverter a DI. Existem alguns relatos que apoiam esta hipótese, no entanto, na maioria dos casos a DI é permanente17.
Conflito de interessesOs autores declaram não haver conflito de interesses.
