



O feocromocitoma é um tumor raro com origem no tecido cromafim. Para avaliar a epidemiologia característica e abordagem destes tumores, foi efetuado um estudo multicêntrico retrospetivo em doentes com feocromocitoma.
Material e métodosEstudo retrospetivo, desenvolvido em 12 centros, incluiu 176 doentes tratados entre 1986‐2011. Efetuado um questionário que incluiu dados epidemiológicos, clínicos, doseamentos laboratoriais, exames de localização, estudo genético, preparação pré‐operatória e cirurgia. De acordo com a data de diagnóstico, os doentes foram divididos em 2 períodos de tempo, 1986‐2000 e 2001‐2011, e alguns dados foram comparados.
ResultadosCento e cinco mulheres e 70 homens, idade média 51,9±15,2 anos. Em 172 doentes, a apresentação clínica foi: 31% incidentalomas, 10% paroxismos típicos, 18% hipertensão persistente e 5% detetados durante rastreio genético. Onze por cento tinham outros sintomas clínicos de feocromocitoma e 25% uma mistura de 2 ou mais quadros clínicos. Os exames laboratoriais mais frequentes foram as catecolaminas urinárias e seus metabolitos urinários.
Em 154 doentes, foi localizado por TAC em 84%, RMN em 41% e a cintigrafia com MIBG em 55%. A dimensão média do tumor foi 55,3±33,7mm, 56% na suprarrenal direita e 7% bilateral. O tratamento pré‐operatório em 126 casos foi: fenoxibenzamina em 65% dos doentes e associada a um betabloqueador em 29,3%.
Em 170 doentes, 91 efetuaram laparotomia (54%) e 74 laparoscopia (44%). Cinco doentes não efetuaram cirurgia. Em 9 doentes foi diagnosticado feocromocitoma maligno, 3 na altura do diagnóstico inicial e 6 durante o seguimento (após 6‐192 meses).
Em 19 doentes foi efetuado o diagnóstico de uma síndrome genética.
ConclusõesTrinta e um por cento dos tumores foram detetados como incidentalomas. A suprarrenal direita foi mais atingida. Observou‐se um aumento do n° de diagnósticos de feocromocitoma e um melhor estudo e estadiamento nos doentes diagnosticados entre 2001 e 2011 comparativamente com os diagnosticados entre 1986 e 2000. Dado que a malignidade se pode manifestar tardiamente, o seguimento destes doentes deve ser para toda a vida.
Pheochromocitoma is a rare tumor arising from the chromaffin tissue. To have an idea of the epidemiology, characteristics and management of this tumors it was performed a multicentric retrospective evaluation of patients with pheochromocitoma.
Material and methodsTwelve endocrine departments participated, reviewed the data of 176 patients, between 1986 and 2011. A questionaire included data on epidemiological, clinical, laboratory, radiological, genetic study, preoperative treatment and surgery. According to year of diagnosis the patients were divided in two groups, 1986‐2000 and 2001‐2011, and some data were compared.
Results105 female and 70 male, with 51.9±15.2 years. The clinical presentation of 172 patients were: 31% discovered incidentally; 10% had the typical spells; 18% had persistent hypertension and 5% discovered in a genetic screening; 11% other clinical symptom of pheochromocytomas and 25% had a mixture of two or more clinical presentation. The most often laboratory assays evaluated were the urinary catecholamines and their urinary metabolites.
In 154 patients, the localization was by CT in 84%, MR in 41% and MIBG scintigraphy in 55%. The mean dimension of the tumor was 55.3±33.7mm, 56% at the right adrenal, and 7% bilateral. The preoperative treatment in 126 cases was: phenoxybenzamine in 65% of the patients, and associated to a β blocker in 29.3%.
In 170 patients, the surgical approach was laparotomy in 91(54%) and laparoscopy in 74(44%); 5 patients had no surgery. 9 patients had the diagnosis of malignant pheochromocitoma: at the diagnosis in 3 cases, or during the follow up (after 6 to 192 months) in 6.
In 19 patients a genetic syndrome was found.
Conclusions31% of the patients discovered as a incidentalomas. The right adrenal was more affected. Between 2001 and 2011 more patients were diagnosed with pheochromocitoma and their sudy and staging were improved comparing with the period between 1986 and 2000. Given that the malignancy in a pheochromocitoma can be a late finding life long follow‐up is mandatory.
O feocromocitoma é um tumor raro, com origem no tecido cromafim da medula da suprarrenal. Tem uma prevalência variável nos diferentes estudos, sendo de cerca de 0,1‐0,6% nos doentes com hipertensão1. Habitualmente são tumores benignos curáveis pela cirurgia; no entanto, se não corretamente diagnosticados e tratados, podem ter uma elevada taxa de mortalidade e morbilidade.
O diagnóstico pode ser difícil devido à raridade do tumor e ao facto da apresentação clínica ser muito variável, fruto de uma secreção episódica e imprevisível de catecolaminas.
O quadro clínico típico, de paroxismos com cefaleias, hipersudorese e palpitações, está presente em apenas 24% dos casos2.
Atualmente, devido à maior utilização dos meios de imagem, os feocromocitomas são frequentemente descobertos acidentalmente, muitas vezes ainda clinicamente silenciosos, podendo nestas circunstâncias ter um excelente prognóstico3.
Devido à heterogeneidade da apresentação clínica, à possível associação a síndromes hereditárias, ao facto da ocorrência rara de malignidade poder ser um evento tardio, e às diferentes abordagens possíveis desta doença, o Grupo de Estudos de Tumores da Suprarrenal da Sociedade Portuguesa de Endocrinologia, Diabetes e Metabolismo decidiu efetuar um estudo multicêntrico retrospetivo, com o objetivo de obter um quadro da epidemiologia e da metodologia de abordagem destes tumores em Portugal.
Material e métodosRealizou‐se um estudo retrospetivo, desenvolvido em 12 centros de endocrinologia, que incluiu doentes tratados entre 1986‐2011. O diagnóstico de feocromocitoma constituiu o único critério de inclusão. Um dos centros incluiu 17 doentes apenas observados na área cirúrgica. Os casos foram identificados após consulta dos processos clínicos, com registo dos dados referentes a: sexo, idade na altura do diagnóstico, história pessoal e familiar, forma de apresentação clínica, doseamentos laboratoriais, exames de localização, estudo genético, preparação pré‐operatória, tipo de cirurgia e dados da anatomia patológica.
Delinearam‐se 5 formas de apresentação inicial: HTA mantida, paroxismos típicos, deteção devido a suspeita clínica de formas familiares, outros sintomas, e tumores detetados acidentalmente. Consideramos, igualmente, a associação de 2 ou mais formas simultâneas de apresentação.
Incluíram‐se os doseamentos urinários de catecolaminas totais, adrenalina, noradrenalina, dopamina, metanefrinas totais e fracionadas e ácido vanilmandélico; assim como os doseamentos plasmáticos de adrenalina, noradrenalina, metanefrinas, normetanefrinas e cromogranina A. Considerámos os resultados apenas qualitativamente, como normais ou elevados.
Avaliaram‐se ainda os métodos de localização do tumor, estudo genético, tipo de preparação pré‐operatória e abordagem cirúrgica utilizada. Com base na data do diagnóstico da doença, os doentes foram divididos em 2 períodos: 1986‐2000 e 2001‐2011. Comparamos alguns dados clínicos, analíticos, métodos de imagem, estudos genéticos e técnicas cirúrgicas entre os 2 períodos
Análise estatísticaOs dados são fornecidos como médias e desvio‐padrão, ou médias e intervalos, de acordo com o apropriado. Para o cálculo das médias, desvio‐padrão e intervalos, foi utilizado o programa Excel®. Para as comparações estatísticas, foi usado o teste t de Student para amostras independentes. Foi considerado como significativo um valor de p<0,05.
ResultadosEpidemiologia e dados clínicosIdentificámos 176 casos doentes, com idades compreendidas entre 17‐90 anos, dos quais 105 (60%) eram mulheres. A idade média na altura do diagnóstico foi de 51,9±15,2 anos.
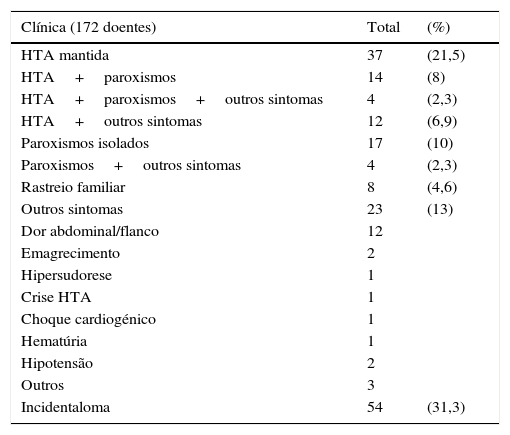
Em 172 casos obteve‐se o registo da forma de apresentação (ver tabela 1). Mais de metade dos doentes apresentava alguma forma de HTA sustentada e/ou associada a paroxismos. Dos 8 doentes (4,6%) cujo diagnóstico foi efetuado devido a suspeita da existência de síndromes genéticas, 5 pertenciam a famílias com neoplasia endócrina multipla tipo 2 A (NEM2A) e 3 apresentavam carcinoma medular da tiroide. Vinte e três doentes (13%) apresentavam outros sintomas, que podiam ou não ser atribuídos ao feocromocitoma.
Forma de apresentação clínica
| Clínica (172 doentes) | Total | (%) |
|---|---|---|
| HTA mantida | 37 | (21,5) |
| HTA+paroxismos | 14 | (8) |
| HTA+paroxismos+outros sintomas | 4 | (2,3) |
| HTA+outros sintomas | 12 | (6,9) |
| Paroxismos isolados | 17 | (10) |
| Paroxismos+outros sintomas | 4 | (2,3) |
| Rastreio familiar | 8 | (4,6) |
| Outros sintomas | 23 | (13) |
| Dor abdominal/flanco | 12 | |
| Emagrecimento | 2 | |
| Hipersudorese | 1 | |
| Crise HTA | 1 | |
| Choque cardiogénico | 1 | |
| Hematúria | 1 | |
| Hipotensão | 2 | |
| Outros | 3 | |
| Incidentaloma | 54 | (31,3) |
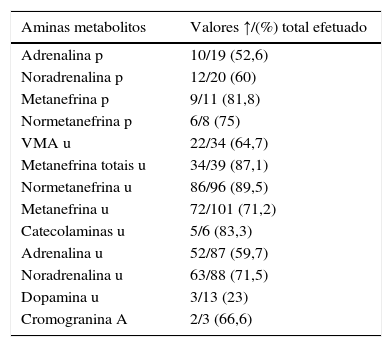
Em 28 doentes (16%) não se obteve informação relativa a dados bioquímicos. Nos 148 restantes (84%), os mais frequentes foram doseamentos de adrenalina, noradrenalina, metanefrinas e normetanefrinas, na urina de 24h (tabela 2). Num subgrupo de 52 doentes, em que os doseamentos foram efetuados por HPLC, com valores de referência semelhantes, comparou‐se a sensibilidade das aminas e respetivos metabolitos para a deteção da doença. O teste mais sensível foi o doseamento de normetanefrina na urina de 24h (tabela 3).
Estudo hormonal pré‐operatório (n=148)
| Aminas metabolitos | Valores ↑/(%) total efetuado |
|---|---|
| Adrenalina p | 10/19 (52,6) |
| Noradrenalina p | 12/20 (60) |
| Metanefrina p | 9/11 (81,8) |
| Normetanefrina p | 6/8 (75) |
| VMA u | 22/34 (64,7) |
| Metanefrina totais u | 34/39 (87,1) |
| Normetanefrina u | 86/96 (89,5) |
| Metanefrina u | 72/101 (71,2) |
| Catecolaminas u | 5/6 (83,3) |
| Adrenalina u | 52/87 (59,7) |
| Noradrenalina u | 63/88 (71,5) |
| Dopamina u | 3/13 (23) |
| Cromogranina A | 2/3 (66,6) |
Na 2.a e 3.a colunas são apresentados, respetivamente, o n.° dos doseamentos elevados e % em relação ao total das determinações.
Obtivemos informação dos estudos de localização em 164 doentes. O exame mais utilizado foi a tomografia axial computorizada (TAC) em 129 doentes (78,6%), seguido da ressonância magnética (RMN) em 63 (38,4%). Cento e quarenta e cinco doentes (88,4%) efetuaram apenas um exame radiológico (TAC ou RMN) e em 28 casos (17%) foram ambos realizados (TAC e RMN). Em 155 doentes (correspondendo a 167 tumores) houve registo das dimensões do tumor no exame radiológico. A média em 167 tumores foi de 54,8±32,6mm (entre 11‐200mm). Comparando a média das dimensões dos tumores identificados com um único exame radiológico – 56,3±34,0mm (11‐200) (em 134 tumores) – com a média das dimensões dos que fizeram ambos os exames – 43,4±19,2mm (11‐100) (em 29 tumores) –, verifica‐se que estes tinham menores dimensões, mas no limite da significância: p=0,05.
Em 84 doentes foi efetuado cintigrama com meta‐iodo‐benzil‐guanidina (MIBG), utilizando 123I ou 131I. Em 6 doentes (7%) não houve fixação do radiofármaco pelo tumor; destes, num dos casos o tumor era bilateral. A dimensão média dos 7 tumores falsos negativos era de 39±20,4mm (20‐80mm). Em 4 doentes (4,7%), além da suprarrenal comprometida, houve também discreta fixação na suprarrenal contra lateral, sem doença.
Em 83 doentes (53,5%) a localização foi na suprarrenal direita, em 57 casos (36,7%) localizavam‐se na suprarrenal esquerda, e em 11 doentes (7%) bilaterais. Os tumores localizados à direita mediam, em média, 53,6±32,1mm e à esquerda 59,8±34,1mm.
Tratamento médico pré‐cirúrgicoEm 126 doentes havia o registo dos fármacos utilizados na preparação pré‐operatória. A duração média do bloqueio dos recetores adrenérgicos antes da cirurgia, conhecida em 101 doentes, foi de 32,4±37,7 (5‐294) dias. A fenoxibenzamina em monoterapia foi utilizada em 82 doentes (65%) e associada a um ß bloqueador em 37 casos (29,3%). Em 3 doentes (2,3%), a fenoxibenzamina foi associada a bloqueador ß e outros anti‐hipertensores. Foram utilizados em 4 doentes (3,1%) antagonistas α1‐seletivos associados a outros anti‐hipertensores.
Tratamento cirúrgicoCento e setenta e dois doentes foram submetidos a cirurgia e obtivemos informação do registo operatório em 166 casos. Setenta e quatro doentes (44%) foram submetidos a laparoscopia e 92 doentes (54%) a laparotomia.
Em 66 tumores abordados por laparoscopia, a média das dimensões foi de 42,8mm (15‐138) e em 94 tumores que efetuaram laparotomia, a média das dimensões foi de 63,2mm (11‐200), sendo a diferença significativa entre os 2 grupos (p=0,0001). Sete doentes efetuaram suprarrenalectomia simultânea de ambas as suprarrenais e 2 casos efetuaram suprarrenalectomia poupadora do córtex. Quatro doentes não realizaram cirurgia: 2 por recusa, um por ter um tumor considerado irressecável e o último por ter falecido antes da cirurgia (enfarte agudo do miocárdio).
HistologiaCento e sessenta e três doentes (93%) tiveram o diagnóstico de feocromocitoma confirmado histologicamente.
Em 9 casos (5,2%) não houve acesso a histologia por terem sido operados noutros hospitais.
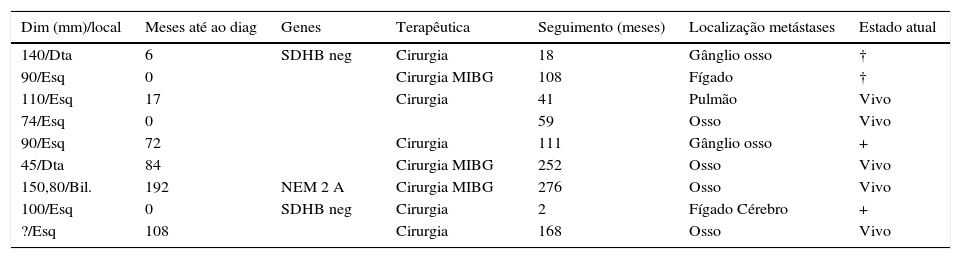
MalignidadeO diagnóstico de malignidade foi efetuado em 9 doentes (5%) (tabela 4), 4 mulheres e 5 homens, com idade média 51,8±16,8 anos (23‐71).
Dados e evolução de 9 doentes com feocromocitoma maligno
| Dim (mm)/local | Meses até ao diag | Genes | Terapêutica | Seguimento (meses) | Localização metástases | Estado atual |
|---|---|---|---|---|---|---|
| 140/Dta | 6 | SDHB neg | Cirurgia | 18 | Gânglio osso | † |
| 90/Esq | 0 | Cirurgia MIBG | 108 | Fígado | † | |
| 110/Esq | 17 | Cirurgia | 41 | Pulmão | Vivo | |
| 74/Esq | 0 | 59 | Osso | Vivo | ||
| 90/Esq | 72 | Cirurgia | 111 | Gânglio osso | + | |
| 45/Dta | 84 | Cirurgia MIBG | 252 | Osso | Vivo | |
| 150,80/Bil. | 192 | NEM 2 A | Cirurgia MIBG | 276 | Osso | Vivo |
| 100/Esq | 0 | SDHB neg | Cirurgia | 2 | Fígado Cérebro | + |
| ?/Esq | 108 | Cirurgia | 168 | Osso | Vivo |
Em 3 casos (33%), a doença metastática foi identificada na altura do diagnóstico do feocromocitoma; nos restantes, o tempo até ao aparecimento de metástases, variou entre 6‐192 meses.
Em 8 doentes (89%), os tumores eram unilaterais. Todos os doentes efetuaram cirurgia, com exceção do doente número 4, que tinha uma massa irressecável.
Três doentes (33%) realizaram uma ou mais sessões terapêuticas com MIBG após a cirurgia.
Quatro doentes morreram devido ao feocromocitoma maligno.
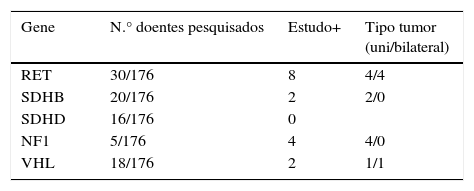
Estudo genéticoTrinta e oito doentes (22%) efetuaram a pesquisa de mutações de um ou mais genes (tabela 5). Com base no estudo genético efetuado e em 2 doentes que já tinham diagnóstico prévio, foram identificados 16 casos com síndromes familiares. Cinco doentes com síndromes genéticas tinham tumores bilaterais, 4 dos quais com NEM tipo 2A.
Atualmente, 13 doentes estão livres de doença, um teve uma recorrência, um está perdido para seguimento e um tem o diagnóstico de malignidade.
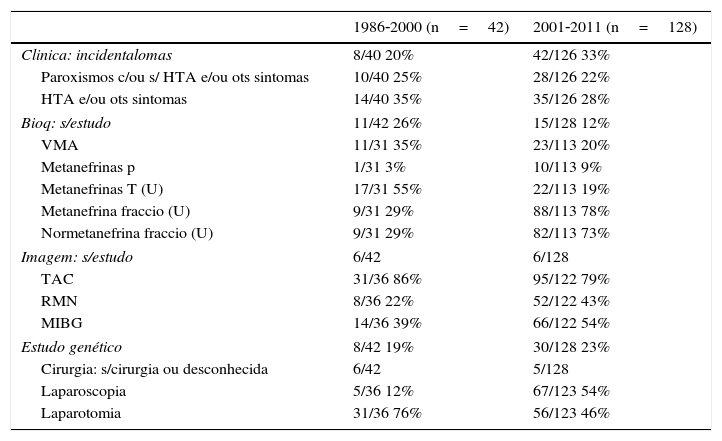
Evolução temporal – ano de diagnóstico do feocromocitomaEm 170 doentes, foi conhecido o ano de diagnóstico da doença. Os doentes foram separados por 2 períodos e alguns dados comparados (tabela 6). Quarenta e dois doentes diagnosticados entre 1986‐2000 e 128 com o diagnóstico entre 2001‐2011.
Características dos doentes de acordo com o período temporal
| 1986‐2000 (n=42) | 2001‐2011 (n=128) | |
|---|---|---|
| Clinica: incidentalomas | 8/40 20% | 42/126 33% |
| Paroxismos c/ou s/ HTA e/ou ots sintomas | 10/40 25% | 28/126 22% |
| HTA e/ou ots sintomas | 14/40 35% | 35/126 28% |
| Bioq: s/estudo | 11/42 26% | 15/128 12% |
| VMA | 11/31 35% | 23/113 20% |
| Metanefrinas p | 1/31 3% | 10/113 9% |
| Metanefrinas T (U) | 17/31 55% | 22/113 19% |
| Metanefrina fraccio (U) | 9/31 29% | 88/113 78% |
| Normetanefrina fraccio (U) | 9/31 29% | 82/113 73% |
| Imagem: s/estudo | 6/42 | 6/128 |
| TAC | 31/36 86% | 95/122 79% |
| RMN | 8/36 22% | 52/122 43% |
| MIBG | 14/36 39% | 66/122 54% |
| Estudo genético | 8/42 19% | 30/128 23% |
| Cirurgia: s/cirurgia ou desconhecida | 6/42 | 5/128 |
| Laparoscopia | 5/36 12% | 67/123 54% |
| Laparotomia | 31/36 76% | 56/123 46% |
Na 2.a e 3.a colunas, o n.° de doentes que apresentava esse dado em relação ao n.° total efetuado e respetivas percentagens.
Constatamos a grande variabilidade na expressão clínica deste tipo de tumores e o facto da apresentação clássica, com sintomatologia paroxística, ser relativamente pouco frequente. Esta realidade tem sido também referida em várias séries na literatura1–4.
Com a crescente sofisticação dos exames de imagem e a sua maior utilização, o diagnóstico de incidentalomas é cada vez mais frequente.
A deteção acidental de massas da suprarrenal depende, sobretudo, da idade do doente e do tipo de exame radiológico5,6; cerca de 5‐7% dos incidentalomas são feocromocitomas7,8. A frequência de feocromocitomas descobertos acidentalmente varia nas diferentes séries entre 11‐57%1,4,9,10. No nosso estudo, 30% dos feocromocitomas foram descobertos acidentalmente. Verificamos também que não só o n.° de diagnósticos de feocromocitoma aumentou no período de 2001‐2011, como também estes foram mais vezes diagnosticados como incidentalomas. Pensamos que a melhoria dos cuidados de saúde em geral, associada a um aumento dos estudos de imagem, é responsável por este facto.
Tal como na experiência de outros centros1,4,9, as maiores limitações do nosso estudo foram as análises laboratoriais. Foram utilizados vários métodos de doseamento (tanto das aminas como dos seus metabolitos) – nos diferentes centros, numa mesma época, e em cada centro, ao longo dos 25 anos em que se estende o estudo. Verificámos também que doseamentos utilizando a mesma técnica laboratorial podiam ter diferentes valores de referência nos diversos hospitais.
Acresce ainda o facto de que, na interpretação de todos estes resultados, devemos ter em conta as várias causas potenciais de interferência11,12, os níveis hormonais obtidos (n.° de vezes o máximo do normal)13,14 e o facto de que os feocromocitomas poderem ter uma secreção episódica.
Todos esses fatores devem ser considerados quando interpretamos os resultados14, traduzindo a frequente dificuldade em estabelecer o diagnóstico definitivo.
Na nossa série, verificou‐se que 28 doentes não tinham qualquer quantificação de aminas. A ausência de qualquer estudo bioquímico foi sobretudo notória quando a doença foi diagnosticada entre 1986‐2000, em que mais de um quarto dos doentes não tinha qualquer estudo. Constatamos igualmente que, apesar um aumento do n.° de pedidos no 2.° período de tempo de diagnóstico, apenas num número muito reduzido de doentes efetuou o doseamento das metanefrinas plasmáticas, considerado hoje como um dos métodos mais sensíveis14–16. No entanto, o pedido dos doseamentos das metanefrinas e normetanefrinas fracionadas na urina de 24h, igualmente com elevada especificidade e sensibilidade diagnóstica, tiveram um aumento marcado quando comparamos o ano de diagnóstico entre 1986‐2000 com 2001‐2011. Os metabolitos urinários das catecolaminas mostraram na nossa série uma sensibilidade inferior ao habitualmente referido17.
A TAC e a RMN são habitualmente altamente sensíveis para a localização destes tumores18,19. Na nossa série, ambos os exames tiveram uma sensibilidade de 100%.
Como seria de esperar devido a um maior acesso a este exame, o estudo por RMN praticamente duplicou quando o diagnóstico foi efetuado entre 2001‐2011.
Um dos motivos para a realização de TAC e RMN em 17% dos doentes poderá ter‐se devido a menor dimensão dos tumores, embora a diferença de tamanho em relação aos tumores localizados por um único exame se situe no limite da significância.
A realização de cintigrafia com MIBG marcada com um isótopo de iodo (I123 ou I131), como avaliação de base em todos os doentes, não é consensual20. Aceita‐se a sua indicação em casos de tumores de grandes dimensões e, consequentemente, maior probabilidade de malignidade, ou nos casos de elevada suspeita de doença multifocal11,14.
O estadiamento da doença pelo MIBG foi efetuado em cerca de um terço dos doentes no período de 1986‐2000 e em mais de metade dos doentes quando o diagnóstico foi efetuado após 2000.
A suprarrenal normal pode captar fisiologicamente a MIBG21. No nosso estudo, constatámos que em 4 doentes houve captação do MIBG pela suprarrenal não afetada.
Os falsos negativos no estudo dos feocromocitomas podem estar relacionados: com uma menor sensibilidade ao MIBG, a utilização concomitante de determinados fármacos22 e as pequenas dimensões dos tumores12. A desdiferenciação das células do tumor com a falta dos grânulos de armazenamento das aminas pode ser uma causa para a menor sensibilidade do MIBG, no caso dos feocromocitomas malignos22.
No nosso estudo, houve 7% de falsos negativos na cintigrafia com MIBG. Não foi possível identificar os doentes que efetuaram 123I ou 131I, pelo que não podemos aferir diferenças de sensibilidade entre os diferentes isótopos21. Em média, os tumores que não fixavam o marcador eram mais pequenos, o que pode justificar parcialmente este facto, mas houve um tumor com 80mm cuja cintigrafia com MIBG foi negativa. A malignidade não foi uma possível explicação para os falsos negativos, já que os 3 doentes com feocromocitoma maligno que efetuaram uma cintigrafia com MIBG pré‐operatória tinham captação do radiofármaco.
Tal como em outras séries, encontrámos uma maior incidência de feocromocitomas à direita, não sendo conhecida nenhuma explicação plausível para este resultado4,23.
A abordagem por via laparoscópica é hoje considerada a técnica de eleição, devido a menor risco e taxa de complicações, mesmo em tumores com mais de 6cm de diâmetro, desde que não sejam invasivos24. Constatamos que, entre nós, esta técnica tem vindo a ser progressivamente realizada, apenas em 12% dos doentes quando o diagnóstico foi efetuado entre 1986‐2000 e em mais de 50% dos casos após o ano 2000; no entanto, as experiências são diferentes em cada hospital. Há centros que, por norma, utilizam a técnica por via laparoscópica, outros que recorrem mais a laparotomia; no cômputo geral, a relação entre abordagens por laparoscopia e por laparotomia na nossa casuística é semelhante (ou mesmo superior) à de outras séries descritas na literatura1,9. Como seria de prever, a abordagem por via laparoscópica foi eleita em tumores de menores dimensões. Em caso de doença familiar, a suprarrenalectomia parcial poupadora do córtex da suprarrenal pode prevenir a deficiência permanente dos glucocorticoides, embora possa aumentar a incidência de recorrência do tumor14–16. Recentemente, uma grande série publicada em 201425 sugere que a suprarrenalectomia poupadora do córtex no contexto de doença familiar não tem maior risco de recorrência com a vantagem de menor risco de insuficiência suprarrenal. Na nossa série, apenas 2 doentes efetuaram este tipo de cirurgia, um dos quais com NEM 2 A.
A incidência de malignidade no feocromocitoma é muito variável nos diferentes estudos, situando‐se entre 3‐36%14,26–30, dependendo dos critérios de referenciação, do tempo de seguimento e da presença de determinadas mutações genéticas. O diagnóstico de malignidade baseia‐se na presença de tecido cromafim em locais onde habitualmente este não se encontra26,27. Este diagnóstico significa uma taxa de sobrevida aos 5 anos, entre os 34 e os 60%14,26. No nosso estudo, 5% dos doentes tinham feocromocitoma maligno; num dos doentes, o diagnóstico foi efetuado 16 anos após a cirurgia. Assim, na ausência de critérios histopatológicos seguros, e dado que a malignidade pode ser um evento tardio, o seguimento destes doentes deve ser indefinido14,23.
Até 2002, considerava‐se que 10% dos feocromocitomas eram hereditários; atualmente, pensa‐se que cerca de 20‐30% serão portadores de formas hereditárias14,30. Esta alteração foi motivada pela descrição, a partir de 2000, de formas familiares ligadas à succinato desidrogenase mitocondrial e, sobretudo, pela deteção de mutações germinativas em doentes com feocromocitomas aparentemente esporádicos e história familiar negativa30.
O rastreio genético, em casos de formas aparentemente esporádicas de feocromocitoma, não é consensual14,30,31; no entanto, não tivemos qualquer dúvida que foi escasso na nossa série: apenas 8 doentes efetuaram algum tipo de estudo genético quando diagnosticados entre 1986‐2000. Apesar do rastreio ter melhorado no 2.° período de tempo, mesmo assim, só 23% dos doentes efetuaram a pesquisa de mutações de um ou mais genes. A data do estudo genético foi em alguns doentes efetuada posteriormente ao diagnóstico, pelo que a comparação entre os 2 períodos pode não ser correta. Detetámos apenas 9% de síndromes familiares. Sem dúvida, um dos principais motivos foi incluir vários doentes diagnosticados anteriormente a 2002, a evolução contínua e muito recente nesta área, e o facto do estudo genético como «standard of care» apenas passar a ser preconizado recentemente. É geralmente referida a maior probabilidade de existirem tumores bilaterais e tumores multifocais nas formas familiares31; nos nossos casos, em 16 doentes com formas genéticas, 5 tinham tumores bilaterais: 4 casos de MEN tipo2 A e um doente com Von Hippel‐Lindau (VHL).
Os casos associados a mutações do SDHB são caracterizados por um alto potencial maligno16,32,33; na nossa série, os 2 doentes identificados com este tipo de mutações mantêm‐se atualmente livres de doença e os 2 doentes com feocromocitoma maligno testados para essa mutação tiveram um resultado negativo.
ConclusãoEste estudo confirma a multiplicidade de apresentação clínica do feocromocitoma e o número significativo de diagnósticos efetuados no decurso de estudos imagiológicos.
Na abordagem destes doentes, é fundamental a utilização de métodos bioquímicos e imagiológicos adequados e sensíveis, de forma a não deixar escapar o diagnóstico de um feocromocitoma.
Dado que a malignidade se pode tornar evidente somente após muitos anos de diagnóstico, o seguimento destes doentes deve ser mantido durante toda a sua vida.
Com a maior facilidade na realização de estudos genéticos e na sua acessibilidade, estes deverão ser propostos a todos os doentes com feocromocitoma esporádico.
Com o maior acesso aos cuidados médicos e o avanço do conhecimento e tecnologia, notamos um aumento do n.° de diagnósticos, e um melhor estudo e estadiamento destes doentes.
Não podemos deixar também de referir a grande dificuldade e as enormes limitações destes estudos retrospetivos: é frequente a falta de dados nos registos e a referência de exames incompletos. Simultaneamente, com a evolução do conhecimento o tipo de estudo analítico e imagiológico vai variando, sendo também dependente da experiência dos diferentes serviços hospitalares.
Este tipo de estudo retrospetivo tem o enorme mérito de mostrar a experiência e a prática nos vários centros, a sua evolução ao longo do tempo, assim como permitir o reconhecimento de lacunas, com as consequentes oportunidades de correção e de atualização dos profissionais envolvidos.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
O Grupo de Estudos da Suprarrenal agradece o apoio prestado à Sociedade Portuguesa de Endocrinologia, Diabetes e Metabolismo.
