




Es bien sabido que el cáncer mamario es considerado un problema de salud a nivel mundial, la enorme tasa de mortalidad se debe a la recaída de la enfermedad, principalmente por la generación de resistencia a los diversos tratamientos. Hasta hace unos años, esta resistencia era atribuida a las mutaciones genéticas heredadas, sin embargo, evidencias recientes sugieren que el microambiente tumoral desempeña un papel clave en el desarrollo y la progresión del cáncer. La relación simbiótica entre las células tumorales y los fibroblastos asociados a cáncer (FAC), condicionan un ambiente propicio para el soporte estructural necesario, lleno de nutrientes que favorecen su crecimiento y progresión. Aquí se describe el papel que juega el microambiente tumoral y los FAC, desde su origen celular y activación, hasta los mecanismos de quimiorresistencia tumoral, además de los cambios epigenéticos y las proteínas involucradas, como las HDAC, que prometen ser blancos terapéuticos de nuevos fármacos dirigidos a su inhibición, al mitigar diversas vías que participan en la activación de los FAC o revertir su potencial promotor de tumores, lo que a su vez, mejoraría la calidad de vida de las pacientes.
It is well known that breast cancer is considered a worldwide health problem, the enormous mortality rate is due to the relapse of patients mostly due to the generation of resistance to various treatments. Until a few years ago, this resistance was attributed to inherited genetic mutations, however, recent evidence suggests that tumor microenvironment plays a key role in the development and progression of cancer. The symbiotic relationship between tumor cells and cancer-associated fibroblasts (CAF) provides an environment conducive to the necessary structural support, full of nutrients that favor their growth and progression. Here we describe the role played by the tumor microenvironment and CAF, from their cellular origin and activation to the mechanisms of tumor chemoresistance, in addition to the epigenetic changes and proteins involved, such as HDAC, which promise to be therapeutic targets for new drugs aimed at their inhibition, by mitigating various pathways involved in the activation of CAF or reversing their tumor-promoting potential, which in turn, would improve the quality of life of patients.
Una neoplasia (o tumor) es una masa de tejido anormal con crecimiento prácticamente autónomo que excede al de los tejidos normales. Las neoplasias pueden ser benignas o malignas y su nomenclatura se basa en las características de su presunta histogénesis. Respecto a los tumores malignos, estos reciben la denominación de cáncer y se dividen en 2 categorías generales: I) carcinomas, que se originan de las células epiteliales; y II) sarcomas, que se originan de las células mesenquimales1. Surgen de una serie de cambios genéticos (mutaciones) y epigenéticos, que usualmente involucran a las proteínas asociadas al ADN que influyen en la expresión génica y dan como resultado la transformación de las células sanas, usualmente de estirpe epitelial. Los cambios en los genes responsables de la carcinogénesis pueden ser heredados o de novo (mutaciones somáticas), los cuales generalmente son producto de la exposición a diversos tipos de estrés ambiental, como la radiación ultravioleta, estilos de vida o la exposición a agentes biológicos como algunos virus2–4.
De acuerdo a COSMIC (Catalogue Of Somatic Mutations In Cancer) más del 1% de todos los genes humanos están implicados en cáncer debido a mutaciones: las mutaciones somáticas están presentes en el 90% de estos genes, 20% presentan mutaciones de la línea germinal, lo que predispone a los portadores a padecer cáncer, y un 10% presentan tanto mutaciones somáticas como de la línea germinal (https://cancer.sanger.ac.uk/cosmic) (mayo 2021). Los diferentes efectos funcionales de los diversos tratamientos, así como el desarrollo del cáncer, pueden deberse a diferentes mutaciones en un mismo gen5.
Cáncer de mamaEl cáncer de mama es considerado una enfermedad de desarrollo y progresión heterogénea6, se mantiene como el segundo lugar en incidencia y mortalidad en las mujeres de todas las edades, con más de 2,2 millones de nuevos casos y más de 680 mil muertes a nivel mundial. En México, este cáncer es considerado un problema grave de salud con casi 30 mil nuevos casos y más de 7 mil muertes en 20207.
Subtipos de cáncer de mamaSe tiene la hipótesis de que los diversos perfiles moleculares y la conducta clínica de los subtipos de cánceres de mama se deben a la plasticidad inherente de las células progenitoras y células madre cancerosas, lo que contribuye al fenotipo tumoral y no el «origen celular» per se8.
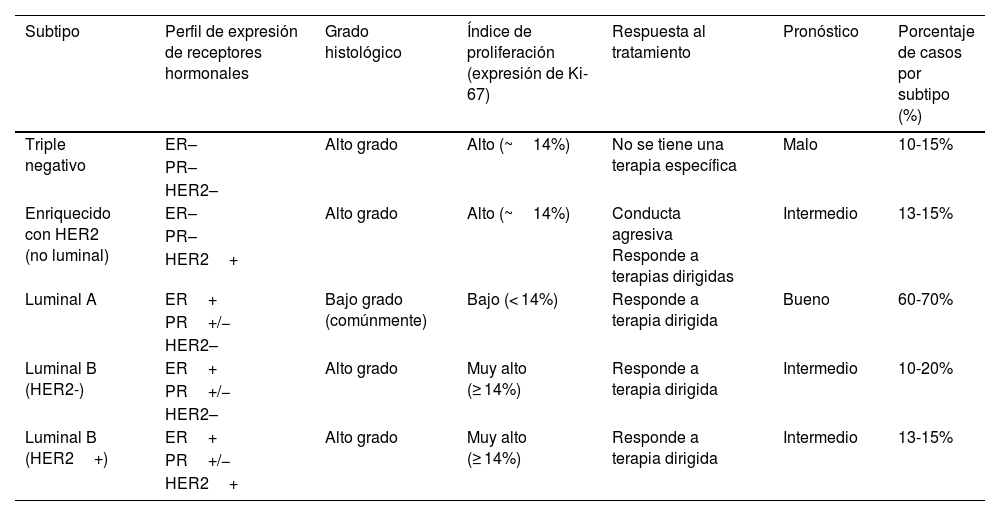
Actualmente, en la clínica es común el uso de la clasificación conocida como «subtipos intrínsecos sustitutos»; engloban 5 subtipos y se basan principalmente, en las diferentes características biológicas del tejido tumoral, como: estatus endocrino, grado histológico (escala Scarff-Bloom-Richardson modificada por Elston y Ellis)9, e índice de proliferación (expresión de Ki-67)10 (tabla 1).
Clasificación intrínseca sustituta
| Subtipo | Perfil de expresión de receptores hormonales | Grado histológico | Índice de proliferación (expresión de Ki-67) | Respuesta al tratamiento | Pronóstico | Porcentaje de casos por subtipo (%) |
|---|---|---|---|---|---|---|
| Triple negativo | ER– | Alto grado | Alto (~14%) | No se tiene una terapia específica | Malo | 10-15% |
| PR– | ||||||
| HER2– | ||||||
| Enriquecido con HER2 (no luminal) | ER– | Alto grado | Alto (~14%) | Conducta agresiva Responde a terapias dirigidas | Intermedio | 13-15% |
| PR– | ||||||
| HER2+ | ||||||
| Luminal A | ER+ | Bajo grado (comúnmente) | Bajo (< 14%) | Responde a terapia dirigida | Bueno | 60-70% |
| PR+/− | ||||||
| HER2– | ||||||
| Luminal B (HER2-) | ER+ | Alto grado | Muy alto (≥ 14%) | Responde a terapia dirigida | Intermedio | 10-20% |
| PR+/− | ||||||
| HER2– | ||||||
| Luminal B (HER2+) | ER+ | Alto grado | Muy alto (≥ 14%) | Responde a terapia dirigida | Intermedio | 13-15% |
| PR+/− | ||||||
| HER2+ |
ER: receptor estrogénico; HER2: receptor tipo 2 al factor de crecimiento epidérmico humano; PR: receptor a progesterona.
Una gran variedad de los tejidos corporales están constituidos por un epitelio que se asocia con un estroma; este comprende una variedad de componentes proteicos de la matriz extracelular (MEC) como proteoglicanos, ácido hialurónico, fibronectina, colágeno y laminina; componentes celulares como adipocitos, fibroblastos, células endoteliales, neuronales e inmunes, además de varios factores de crecimiento, citocinas, quimiocinas, anticuerpos y metabolitos11–14.
La organización de una glándula mamaria adulta conserva la estratificación de las capas celulares y MEC en la arborización de los lóbulos y los ductos que componen la mama. La capa de células mioepiteliales que rodean el epitelio mamario, compuesto por las células productoras de leche y las células luminales, es la responsable de crear y mantener la membrana basal; estructura extremadamente densa compuesta por proteoglicanos, colágena IV, lamininas, fibronectina y tenascina, rodeada por una MEC estromal compuesta principalmente por colágeno I15. La MEC además de dar soporte estructural, funciona como un puente en la comunicación entre el epitelio y su microambiente durante el desarrollo del tejido16,17.
Las alteraciones histológicas estromales y en el tejido adyacente a las lesiones malignas, conocida como desmoplasia, puede presentar características diversas como un estroma predominantemente celular y poca matriz, hasta un mínimo contenido celular y un máximo de matriz18,19.
La relación entre la biología estromal, el desarrollo y la progresión tumoral fue establecida por Boyd et al. (2002), quienes encontraron un aumento de 2-6 veces en la susceptibilidad de padecer cáncer de mama en las mujeres con una elevada densidad mamaria (mamografía), y altos niveles de IGF-1 y TIMP-320. Martín y Boyd (2008) relacionaron además, el aumento en la densidad mamográfica con un aumento en el número de células y la deposición de colágeno en el estroma mamario, siendo más significativa la correlación con la deposición de colágeno21. También se ha demostrado que los tumores surgen más frecuentemente en las zonas de mayor densidad en los tejidos mamarios heterogéneos22,23.
Funciones de la MEC en cáncerLa migración celular a través de las matrices fibrilares, como colágeno 1, se da de forma cíclica; la rotura proteolítica de la MEC y los cambios morfológicos de las células mediante la formación de protuberancias y adhesiones focalizadas permiten la contracción y el desprendimiento de la matriz, mientras la célula avanza24. Grzincic y Murphy (2015) mostraron que cambios en la morfología celular, la migración, la expresión de proteasas y la difusión molecular eran alteradas al modificar la estructura y las propiedades mecánicas de una matriz 3D de colágeno 1 con el uso de nanovarillas de oro, promoviendo la migración celular25.
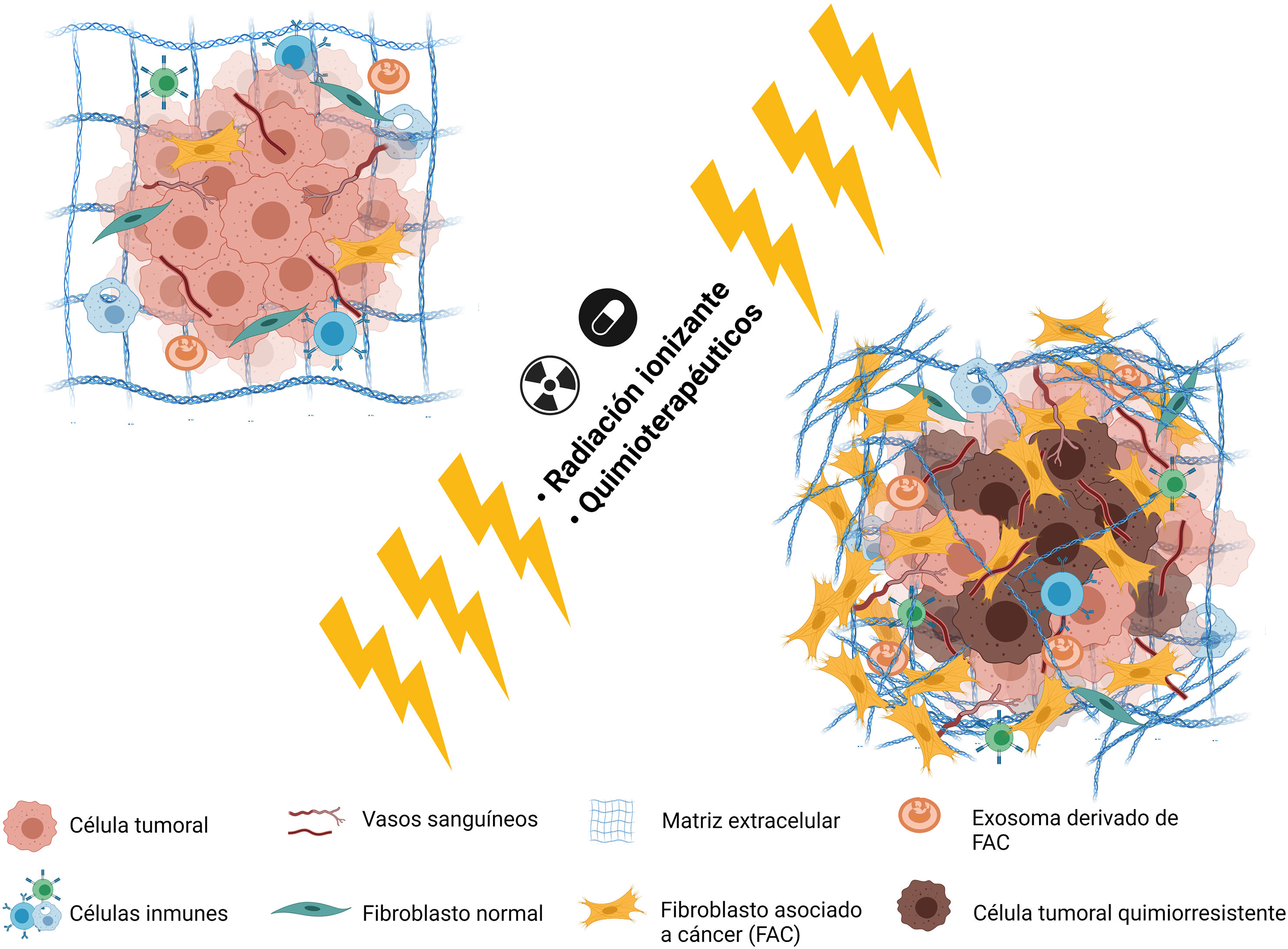
Se ha relacionado el depósito de colágeno con diversas características de tumores mamarios malignos avanzados como la resistencia al tamoxifeno. Jansen et al. (2005) encontraron que, de 81 genes relacionados con la resistencia farmacológica en los pacientes de carcinoma mamario avanzado, 9% estaban relacionados con la formación de la MEC, incluido el gen COLA1A26. Los niveles de MMP-9 se incrementan por la colágena 1 secretada por los miofibroblastos estromales, confiriendo mayor agresividad a las células27 (fig. 1).

Los cambios químicos y mecánicos en el microambiente tumoral (MET), afectan en primera instancia al metabolismo celular28. A pesar de que la influencia del MET en el metabolismo celular aún no ha sido esclarecida por completo, diversos estudios indican que los cambios estromales pueden aumentar el metabolismo, favoreciendo la metástasis y la proliferación celular mediante alteraciones en la densidad del colágeno en la MEC. Morris et al. (2016) realizaron un estudio en las líneas celulares de carcinoma mamario, encontrando que una mayor densidad de colágeno favorece una morfología celular aberrante y una reprogramación del metabolismo, permitiendo una adaptación celular a su entorno, sugiriendo la posibilidad de que la matriz extracelular es internalizada y degradada para funcionar como combustible en el ciclo de los ácidos tricarboxílicos29.
Fibroblastos asociados a cáncer en la mamaLos fibroblastos son las células estromales más abundantes, su función en los tejidos normales del adulto, es el depósito y remodelación de la MEC, produciendo diversos componentes de la misma y proteasas, que ayudan en la remodelación de la matriz12,30. Los fibroblastos activados conocidos como miofibroblastos son una población celular heterogénea y principal representante de FAC, cuya morfología es delgada, cruciforme alargada o en forma de estrella; comparado con los fibroblastos normales, poseen un mayor tamaño, el núcleo dentado, el citoplasma más basófilo y numerosas fibras de tensión31–34; su función depende del tipo de tumor12,35. Se ha visto una relación entre la actividad de los FAC y el crecimiento e invasibilidad de las células cancerosas, en la angiogénesis, así como una disminución de la apoptosis en las células epiteliales adyacentes12,36–38. Las células tumorales son capaces de producir factores que reclutan y activan FAC. Los FAC y los fibroblastos que sobreexpresan el factor de crecimiento transformante beta (TGF-β) promueven la iniciación tumoral en los modelos experimentales. TGF-β participa en la activación y en la actividad de los fibroblastos12,39,40.
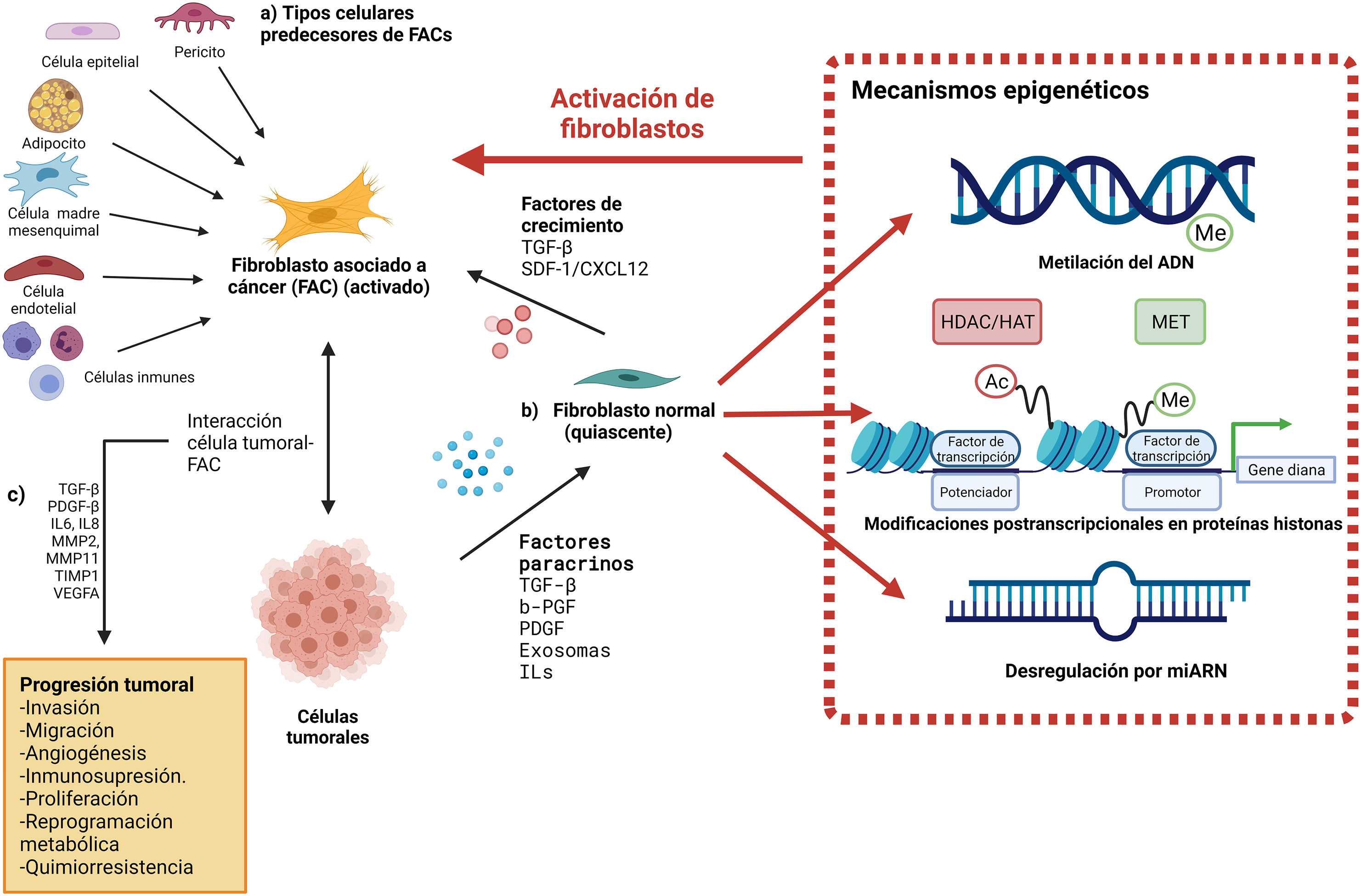
Aunque su mecanismo no ha sido descrito por completo, se sabe que los FAC pueden derivarse de diversos tipos celulares como: fibroblastos normales, adipocitos, células madre mesenquimales, pericitos, entre otros41–43. Aproximadamente el 80% de los fibroblastos normales se transforman en fibroblastos asociados al cáncer de mama (FACM), aunque pueden derivarse de otros precursores12,42,44 (fig. 2A).
 Los FAC pueden derivarse de diferentes precursores como son células epiteliales, células de músculo liso vascular, células endoteliales, entre otras. B) Su activación coordinada y dirigida por activadores y represores transcripcionales, microARN y enzimas que participan en las modificaciones postranscripcionales que reprograman la epigenética del genoma (MET, HDAC y HAT). C) Una vez activados, los FAC permiten la progresión tumoral por diversos mecanismos al aumentar la concentración de ciertos factores y proteínas en la MEC. Creada con BioRender.com.")
El origen y los mecanismos de activación de los FAC. A) Los FAC pueden derivarse de diferentes precursores como son células epiteliales, células de músculo liso vascular, células endoteliales, entre otras. B) Su activación coordinada y dirigida por activadores y represores transcripcionales, microARN y enzimas que participan en las modificaciones postranscripcionales que reprograman la epigenética del genoma (MET, HDAC y HAT). C) Una vez activados, los FAC permiten la progresión tumoral por diversos mecanismos al aumentar la concentración de ciertos factores y proteínas en la MEC. Creada con BioRender.com.
El cambio fenotípico de fibroblastos normales a FAC son procesos coordinados y dirigidos por activadores y represores transcripcionales, microARN y enzimas que participan en las modificaciones postranscripcionales que reprograman la epigenética del genoma, especialmente las histonas-desacetilasas (HDAC)45. Las HDAC son responsables de retirar los grupos acetilo de los residuos de lisina en las proteínas histonas reprimiendo la transcripción de los genes mediante la condensación de la cromatina; mientras que su contraparte, las histona-acetiltransferasas (HAT) agregan grupos acetilo, permitiendo la transcripción genética. Los cambios epigenéticos reversibles, ya sea por la actividad de las HDAC, HAT o por la metilación del ADN pueden ser transmitidos de las células madre a las hijas46–48. Se han identificado diferentes isotipos de las HDAC, las cuales se dividen en 4 clases49. La clase I (HDAC1, 2, 3 y 8), IIa (HDAC4, 5, 7 y 9), IIb (HDAC6 y 10) y IV (HDAC11) son enzimas dependientes de Zn2+ y la clase III (sirtuinas, SIRT1-7) es dependiente de NAD50 (fig. 2B).
El proceso de formación de los FACM comprende 3 pasos: 1) el reclutamiento de las células distantes por células cancerosas; 2) la reprogramación y transformación de las células precursoras sanas a FACM mediante la señalización paracrina; y 3) el mantenimiento constitutivo del fenotipo pro-tumorigénico en la transición epitelio-mesénquima (TEM) de los fibroblastos activados; y el consecuente mantenimiento de la progresión tumoral51.
Para que un FACM adquiera la capacidad pro-tumoral, requiere cambiar su perfil de expresión génica, estas alteraciones pueden darse por cambios epigenéticos52,53 o alteraciones genéticas54,55. Bauer et al. (2010) identificaron la modificación de 22 genes relacionados a la migración y adhesión celular, la regulación de la transcripción y las vías de señalización paracrina e intercelular56; además, se ha encontrado que los FACM carecen de expresión de los supresores tumorales como p21, p53, y de proteínas como PTEN42,44,57.
La actividad de los FACM tiene diferentes efectos en la tumorigénesis mamaria como regular el metabolismo, reprogramar las células tumorales y remodelar la MEC. Se sabe que todos estos eventos tienen un efecto negativo en el pronóstico del paciente, al aumentar la proliferación de las células malignas, su capacidad invasiva y de metástasis44,58–60. Otra evidencia de que algunos FACM participan activamente en el desarrollo y la progresión tumoral, es la diversidad de compuestos presentes en co-cultivos de células de carcinoma mamario y FACM, los cuales promueven la proliferación, la angiogénesis, la metástasis, la resistencia farmacológica y la invasibilidad celular, por ejemplo un aumento de TGF-β, PDGF-β, interleucinas 6 y 8, metaloproteinasas de matriz (MMP2 y MMP11), inhibidor tisular de MMP1 (TIMP1), factor de crecimiento del endotelio vascular A (VEGFA), entre otros, los cuales no se encuentran en monocultivos de células cancerosas61,62 (fig. 2C).
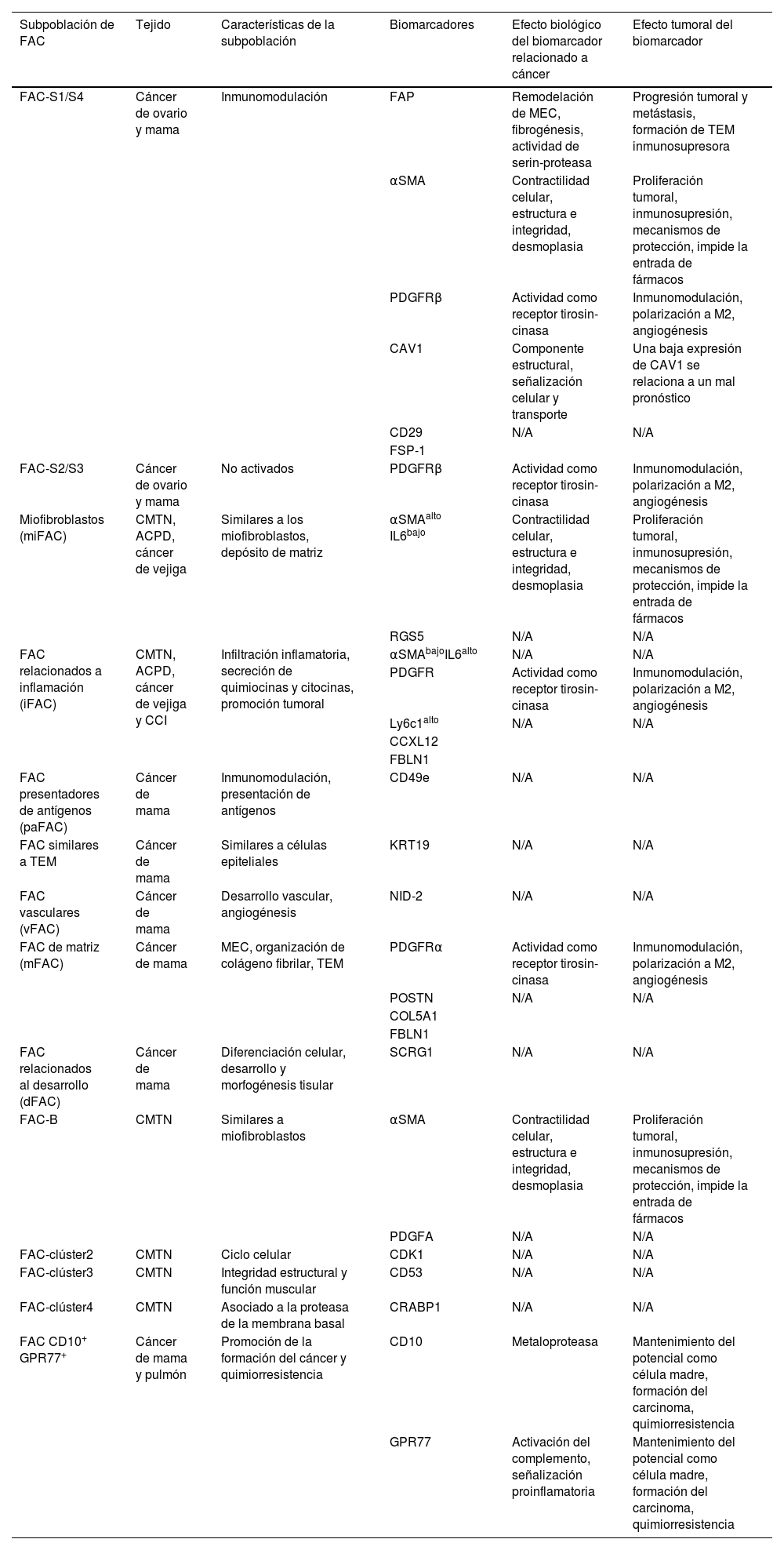
Anteriormente, se consideraba a los FACM como células protumorogénicas, sin embargo, nuevas evidencias sugieren que su heterogeneidad, la cual puede ser resultado de sus diferentes precursores, y del efecto del MET específico de cada carcinoma, provee tanto propiedades promotoras de tumores como antitumorales63,64. Diversos estudios demuestran la diversidad de FAC, dichas células presentan funciones específicas e influencian la respuesta a los tratamientos de las células tumorales. La identificación de marcadores de superficie de los FAC permite manipular de manera específica subpoblaciones de estas células con el fin de lograr un objetivo concreto durante el tratamiento contra el cáncer64–67.
A continuación, se describen las diversas subpoblaciones de FACM asociadas a CM, la expresión de biomarcadores característicos, así como sus funciones biológicas y protumorales64 (tabla 2).
Subpoblaciones de FAC y sus diversos biomarcadores
| Subpoblación de FAC | Tejido | Características de la subpoblación | Biomarcadores | Efecto biológico del biomarcador relacionado a cáncer | Efecto tumoral del biomarcador |
|---|---|---|---|---|---|
| FAC-S1/S4 | Cáncer de ovario y mama | Inmunomodulación | FAP | Remodelación de MEC, fibrogénesis, actividad de serin-proteasa | Progresión tumoral y metástasis, formación de TEM inmunosupresora |
| αSMA | Contractilidad celular, estructura e integridad, desmoplasia | Proliferación tumoral, inmunosupresión, mecanismos de protección, impide la entrada de fármacos | |||
| PDGFRβ | Actividad como receptor tirosin-cinasa | Inmunomodulación, polarización a M2, angiogénesis | |||
| CAV1 | Componente estructural, señalización celular y transporte | Una baja expresión de CAV1 se relaciona a un mal pronóstico | |||
| CD29 | N/A | N/A | |||
| FSP-1 | |||||
| FAC-S2/S3 | Cáncer de ovario y mama | No activados | PDGFRβ | Actividad como receptor tirosin-cinasa | Inmunomodulación, polarización a M2, angiogénesis |
| Miofibroblastos (miFAC) | CMTN, ACPD, cáncer de vejiga | Similares a los miofibroblastos, depósito de matriz | αSMAalto IL6bajo | Contractilidad celular, estructura e integridad, desmoplasia | Proliferación tumoral, inmunosupresión, mecanismos de protección, impide la entrada de fármacos |
| RGS5 | N/A | N/A | |||
| FAC relacionados a inflamación (iFAC) | CMTN, ACPD, cáncer de vejiga y CCI | Infiltración inflamatoria, secreción de quimiocinas y citocinas, promoción tumoral | αSMAbajoIL6alto | N/A | N/A |
| PDGFR | Actividad como receptor tirosin-cinasa | Inmunomodulación, polarización a M2, angiogénesis | |||
| Ly6c1alto | N/A | N/A | |||
| CCXL12 | |||||
| FBLN1 | |||||
| FAC presentadores de antígenos (paFAC) | Cáncer de mama | Inmunomodulación, presentación de antígenos | CD49e | N/A | N/A |
| FAC similares a TEM | Cáncer de mama | Similares a células epiteliales | KRT19 | N/A | N/A |
| FAC vasculares (vFAC) | Cáncer de mama | Desarrollo vascular, angiogénesis | NID-2 | N/A | N/A |
| FAC de matriz (mFAC) | Cáncer de mama | MEC, organización de colágeno fibrilar, TEM | PDGFRα | Actividad como receptor tirosin-cinasa | Inmunomodulación, polarización a M2, angiogénesis |
| POSTN | N/A | N/A | |||
| COL5A1 | |||||
| FBLN1 | |||||
| FAC relacionados al desarrollo (dFAC) | Cáncer de mama | Diferenciación celular, desarrollo y morfogénesis tisular | SCRG1 | N/A | N/A |
| FAC-B | CMTN | Similares a miofibroblastos | αSMA | Contractilidad celular, estructura e integridad, desmoplasia | Proliferación tumoral, inmunosupresión, mecanismos de protección, impide la entrada de fármacos |
| PDGFA | N/A | N/A | |||
| FAC-clúster2 | CMTN | Ciclo celular | CDK1 | N/A | N/A |
| FAC-clúster3 | CMTN | Integridad estructural y función muscular | CD53 | N/A | N/A |
| FAC-clúster4 | CMTN | Asociado a la proteasa de la membrana basal | CRABP1 | N/A | N/A |
| FAC CD10+ GPR77+ | Cáncer de mama y pulmón | Promoción de la formación del cáncer y quimiorresistencia | CD10 | Metaloproteasa | Mantenimiento del potencial como célula madre, formación del carcinoma, quimiorresistencia |
| GPR77 | Activación del complemento, señalización proinflamatoria | Mantenimiento del potencial como célula madre, formación del carcinoma, quimiorresistencia |
ACPD: adenocarcinoma pancreático ductal; CAV1: caveolina 1; CD10: antígeno común de la leucemia linfoblástica aguda; CD29: clúster de diferenciación 29; CD49e: integrina alpha 5; CD53: antígeno de superficie de leucocitos CD53; CFB: factor B del complemento; CDK1: cinasa dependiente de ciclina 1; CMTN: cáncer de mama triple negativo; CRABP1: proteína de unión al ácido retinoico 1; CXCL12: quimiocina 12 con motivo C-X-C; C1QA/B/C: subunidad A/B/C del subcomponente C1q del complemento; FAP: proteína de activación de fibroblastos; FBLN1, fibulina-1; FSP1: proteína específica de fibroblasto 1; GPR77: receptor 2 al componente del complemento 5a; IL6: interleucina-6; IL-10: interleucina 10; KRT19: queratina19; Ly6c1: antígeno linfocitario 6c1; MEC: matriz extracelular; PDGFA: factor de crecimiento derivado de plaquetas A; PDGFRα/β: receptor a factor de crecimiento derivado de plaquetas alpha/beta; POSTN: periostina; RGS5: regulador de la señalización de la proteína G 5; SCRG1: estimulador de condrogénesis 1; TEM: transición epitelio-mesénquima: αSMA: alpha actina de músculo liso.
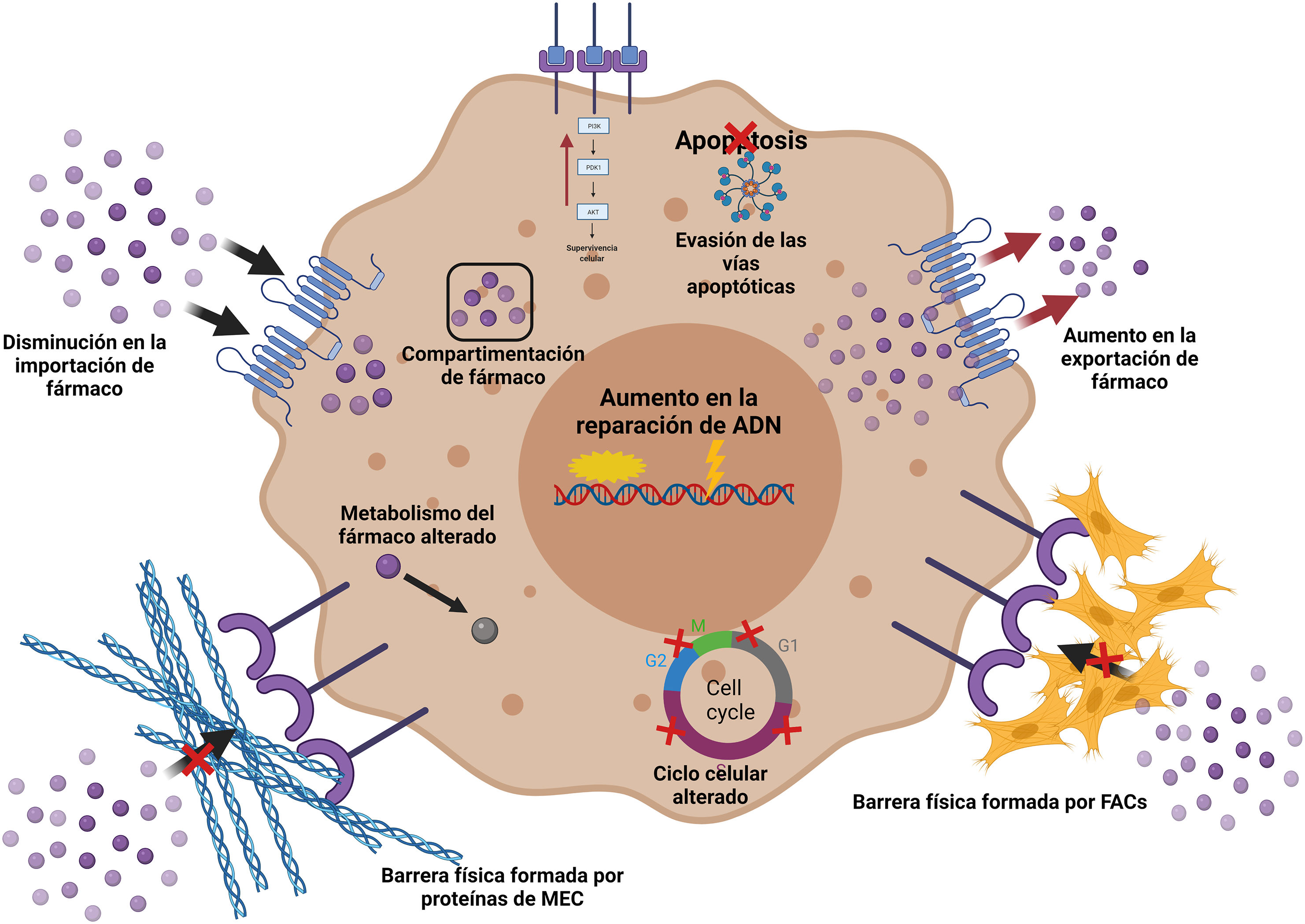
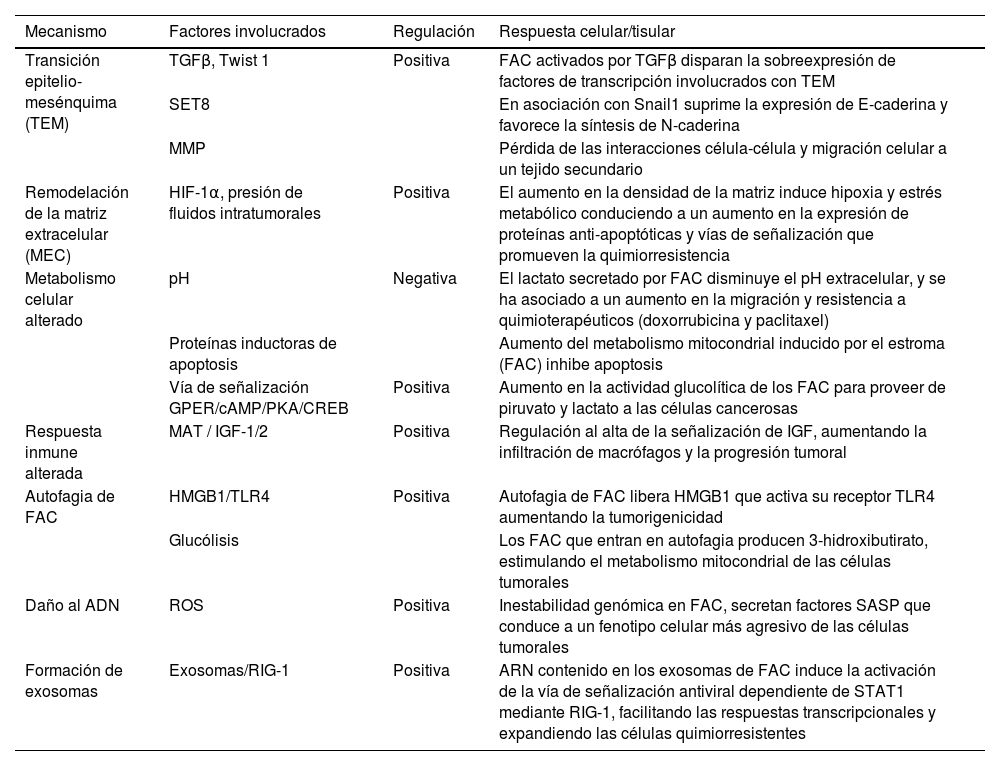
Si bien la resistencia quimioterapéutica se relacionaba exclusivamente a los factores genéticos68, nuevos hallazgos la relacionan con los tejidos adyacentes, al MET y a la cantidad de células estromales activadas, especialmente los FAC69. Una gran variedad de mecanismos, directos e indirectos, se han asociado a la generación de la resistencia farmacológica, desde modificaciones genéticas y epigenéticas, diversidad celular del tumor, metabolismo alterado, aumento en la reparación del ADN, inclusive cambios en el estroma tumoral70,71. La participación de los FAC en la resistencia farmacológica en el carcinoma mamario puede darse por diferentes mecanismos: la inducción de la TEM con la expresión de factores de transcripción (como Snail1/2, Twist1, Zeb1/2) y marcadores (N-caderina, vimentina, αSMA, fibronectina, etc.)72–75; el aumento en la densidad y rigidez de la MEC por proteínas derivadas de FAC, formando una barrera física que impide la entrada de quimioterapéuticos, lo que además dificulta la difusión de oxígeno, nutrientes y metabolitos76,77 y la reprogramación metabólica mediada por FAC78–80 (fig. 3).

Mecanismos implicados en la resistencia a fármacos. Los FAC interactúan con el tumor permitiendo el desarrollo de algunos mecanismos de resistencia: creación de una MEC más densa fungiendo como barrera física; cambios en la importación y la exportación de fármacos, alteraciones en el metabolismo de los fármacos e inhibición de la apoptosis, entre otros. Creada con BioRender.com.
La resistencia a la quimioterapia puede ser inducida también por alteraciones en la respuesta inmune generada por los macrófagos asociados al cáncer y FAC81. Otro mecanismo inductor de resistencia es el mediado por autofagia de los FAC vía HMGB182 o estimulando la glucólisis83. El daño al ADN por la generación de especies reactivas de oxígeno induce una inestabilidad genómica que favorece el desarrollo del cáncer, así como la progresión mediante la resistencia71,84. El potencial intrínseco de transportar material genético y no genético de los exosomas de los FAC a las células tumorales, le brinda un papel importante en la generación de la resistencia a los tratamientos71,85 (tabla 3).
Mecanismos de inducción de resistencia a quimioterapéuticos mediada por FAC
| Mecanismo | Factores involucrados | Regulación | Respuesta celular/tisular |
|---|---|---|---|
| Transición epitelio-mesénquima (TEM) | TGFβ, Twist 1 | Positiva | FAC activados por TGFβ disparan la sobreexpresión de factores de transcripción involucrados con TEM |
| SET8 | En asociación con Snail1 suprime la expresión de E-caderina y favorece la síntesis de N-caderina | ||
| MMP | Pérdida de las interacciones célula-célula y migración celular a un tejido secundario | ||
| Remodelación de la matriz extracelular (MEC) | HIF-1α, presión de fluidos intratumorales | Positiva | El aumento en la densidad de la matriz induce hipoxia y estrés metabólico conduciendo a un aumento en la expresión de proteínas anti-apoptóticas y vías de señalización que promueven la quimiorresistencia |
| Metabolismo celular alterado | pH | Negativa | El lactato secretado por FAC disminuye el pH extracelular, y se ha asociado a un aumento en la migración y resistencia a quimioterapéuticos (doxorrubicina y paclitaxel) |
| Proteínas inductoras de apoptosis | Aumento del metabolismo mitocondrial inducido por el estroma (FAC) inhibe apoptosis | ||
| Vía de señalización GPER/cAMP/PKA/CREB | Positiva | Aumento en la actividad glucolítica de los FAC para proveer de piruvato y lactato a las células cancerosas | |
| Respuesta inmune alterada | MAT / IGF-1/2 | Positiva | Regulación al alta de la señalización de IGF, aumentando la infiltración de macrófagos y la progresión tumoral |
| Autofagia de FAC | HMGB1/TLR4 | Positiva | Autofagia de FAC libera HMGB1 que activa su receptor TLR4 aumentando la tumorigenicidad |
| Glucólisis | Los FAC que entran en autofagia producen 3-hidroxibutirato, estimulando el metabolismo mitocondrial de las células tumorales | ||
| Daño al ADN | ROS | Positiva | Inestabilidad genómica en FAC, secretan factores SASP que conduce a un fenotipo celular más agresivo de las células tumorales |
| Formación de exosomas | Exosomas/RIG-1 | Positiva | ARN contenido en los exosomas de FAC induce la activación de la vía de señalización antiviral dependiente de STAT1 mediante RIG-1, facilitando las respuestas transcripcionales y expandiendo las células quimiorresistentes |
cAMP: adenosín monofosfato cíclico; CREB: elemento de unión de respuesta a cAMP; GPER: receptor estrogénico acoplado a proteínas G; HIF-1α: factor inducible por hipoxia 1α; HMGB1: High Mobility Group Box 1 (proteína no histona asociada a cromatina); IGF1/2: factor de crecimiento similar a insulina 1/2; MAT: macrófagos asociados a cáncer; MMP: metaloproteinasas de matriz; PKA: proteínquinasa A; RIG-1: gen 1 inducido por ácido retinoico antiviral; ROS: especies reactivas de oxígeno; SASP: fenotipo secretor asociado a la senescencia; SET8: histona-metiltransferasa; STAT1: transductor de señales y activado de transcripción. TGFβ: factor de crecimiento transformante beta; TLR4: receptor tipo Toll 4; Twist1: factor de transcripción de TEM.
Debido al importante papel que desempeñan los FAC en la resistencia a la quimioterapia, es posible que una terapia que combine como blancos terapéuticos a los FAC y a las células cancerosas, ofrezca soluciones más eficaces y duraderas para el tratamiento de diversos tipos de cáncer64. Se ha estudiado la posibilidad de diversas terapias contra la activación y actividad de los FAC, como el ácido transretinoico, el cual induce la quiescencia de los FAC o la activación de fibroblastos/células estromales previniendo la secreción aberrante de los factores de crecimiento y citocinas; o utilizando los FAC como transporte, aprovechando su capacidad de almacenaje y exosomas, para acarrear fármacos, virus anticancerígenos o ligandos pro-apoptóticos a las células tumorales76,86–89. La ablación de células estromales como tratamiento contra el cáncer presenta diversos inconvenientes, siendo el más importante, la falta de biomarcadores específicos que identifiquen a los FAC64.
Es bien sabido que las HDAC se sobreexpresan en diversos tipos de cáncer, ya sean sanguíneos o tumores sólidos90 y que los inhibidores de algunas isoformas de estas enzimas (iHDAC) y otras drogas que modifican la epigenética prometen convertirse en nuevos blancos terapéuticos para evitar la progresión de los cánceres con menores efectos adversos. Algunos nuevos tratamientos están diseñados para atacar blancos epigenéticos que contribuyen a la proliferación y la supervivencia de las células cancerosas, sin embargo, pocos estudios se han enfocado en el efecto de la inhibición de las HDAC sobre los subtipos celulares presentes en la MEC y que son de importancia farmacológica debido a sus múltiples efectos, como son los FAC91–97.
Un estudio realizado por Kim et al. (2018), muestra que la actividad in vitro del Scriptaid, un iHDAC, antagoniza la expresión genética mediada por TGFβ en FAC, reduciendo el depósito de proteínas de matriz extracelular, así como su contractilidad y rigidez, además de retrasar el crecimiento tumoral en un modelo in vivo. Potencialmente, el Scriptaid puede revertir el microambiente tumoral que favorece la progresión de la enfermedad al prevenir los procesos mecanosensoriales intrínsecos en los FAC y células tumorales91. Aunque estos nuevos blancos terapéuticos resultan promisorios, se requiere conocer más el papel que desempeñan los diferentes isotipos de estas enzimas en la progresión tumoral, ya que se ha sugerido que algunos iHDAC, especialmente de la isoforma HDAC2, pueden incrementar la expresión de factores que fomentan la progresión del tumor98,99.
El vorinostat (pan-iHDAC) en células 4 T1, inhibe la actividad de la metaloproteinasa de matriz 9 (MMP-9) y reprime la migración e invasibilidad celular. MMP-9 participa en la degradación de colágeno IV y su sobreexpresión se ha asociado con la capacidad invasiva y metastásica en diversos tipos de carcinoma100–104. El uso de vorinostat en combinación con la radiación ionizante ha mostrado que inhibe el crecimiento tumoral en un modelo de carcinoma mamario ortotópico en ratones, indicando una posible sensibilización tumoral a la radioterapia, además de una disminución significativa de la metástasis pulmonar, probablemente por la inhibición de la actividad de la MMP-9105.
Conclusiones y perspectivasCada vez se conoce más sobre el importante papel que juegan la matriz extracelular y los FAC en el desarrollo y la progresión del cáncer, especialmente en la generación de la quimiorresistencia mediante diversos mecanismos como la inducción de la TEM72–75; los cambios en la densidad y la rigidez de MEC por actividad de los FAC76,77, así como la alteración del metabolismo de las células tumorales78–80, entre otras; siendo la adaptación de las células tumorales la causa de las recaídas y la muerte de la mayoría de los pacientes de cáncer. Este nuevo conocimiento, aunado a las herramientas para la generación de nuevos compuestos, como el diseño «in silico» de moléculas basado en la estructura específica de los blancos terapéuticos, como los iHDAC, favorecerá el desarrollo de nuevos tratamientos enfocados a la inhibición de la activación y/o actividad de los FAC y cambios epigenéticos tumorales, los cuales pueden ser usados como monoterapia o en combinación con otros tratamientos. Esta perspectiva es de suma importancia por la posibilidad de tratar enfermedades como el cáncer mamario triple negativo, que carece de blancos farmacológicos de primera elección106 o, utilizarse en tumores malignos resistentes a múltiples terapias.
AgradecimientosAl Dr. José Correa Basurto (ESM-IPN) y al Dr. Javier Ventura Juárez (Ciencias Básicas, UAA) por el apoyo, tiempo y conocimientos aportados para este proyecto.
Consideraciones éticasDicho trabajo no implicó el uso de sujetos humanos o animales.
FinanciaciónEl presente trabajo ha sido financiado por el Consejo Nacional de Ciencia y Tecnología, CONACYT, México, [beca número 761605 (CGRF)], sin participación en la realización de este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

