




El cáncer de mama HER2 positivo supone aproximadamente un 15% de los tumores malignos de mama. La terapia anti-HER2, principalmente representada por trastuzumab, es el pilar del tratamiento de esta enfermedad. La prolongación en la supervivencia que han conseguido las nuevas terapias anti-HER2, y el tratamiento dilatado de estas pacientes en los estadios precoces de la enfermedad así como en la enfermedad avanzada, supone un importante reto a los recursos existentes, tanto con respecto al personal sanitario y el tiempo de los hospitales de día oncológicos, como en cuanto al tiempo dedicado por las pacientes para su tratamiento. El desarrollo de una formulación subcutánea ha supuesto una disminución significativa en la utilización de estos recursos y facilidad de uso. En esta revisión, presentamos el desarrollo de esta nueva formulación, así como los datos de eficacia y preferencia.
HER2-positive breast cancer accounts for approximately 15% of all malignant breast tumors. HER2-targeted therapy, mainly trastuzumab, remains the cornerstone of treatment. The increase in survival achieved by the new anti-HER2 therapies, and the prolonged treatment of these patients, both in the early and advanced breast cancer setting, poses a major challenge to existing health resources. This challenge involves the health personnel, the time consumed by oncologic day hospitals, and the time investment required by patients. The development of the new trastuzumab subcutaneous formulation has improved optimization of health resources and has increased ease of use. In the present article, we review the development of the drug, as well as efficacy and preference data.
El cáncer de mama HER2 positivo supone aproximadamente un 15% de los tumores de mama diagnosticados, y esta característica les confiere mayor agresividad y un peor pronóstico. El tratamiento con fármacos anti-HER2, especialmente trastuzumab (Herceptin®), asociado a la quimioterapia, ha demostrado en numerosos estudios entre un 33 y 50% un 33-50% menos de recaídas en la enfermedad localizada o localmente avanzada de las que de otra forma se producirían sin este tratamiento. El estudio randomizado HERA, en el que participaron 5.102 pacientes y cuyo objetivo principal era determinar la eficacia y seguridad del tratamiento con trastuzumab en la situación adyuvante acompañando a quimioterapia basada en antraciclinas y taxanos, demostró un aumento significativo en la supervivencia libre de enfermedad (SLE; HR 0,64, IC 95% 0,54-0,76; p<0,0001) y en la supervivencia global (SG; HR 0,66, IC 95% 0,47-0,91; p<0,0115) en las pacientes tratadas durante un año con trastuzumab1. Este aumento se ha mantenido tras 8 años de seguimiento2. Los ensayos clínicos NSABP B-31 y NCCTG N9831 arrojaron similares resultados en términos de SLE y SG, también confirmados en el seguimiento a 10 años3,4. Adicionalmente, el estudio BCIRG006 confirmó que esta mejora en la supervivencia podría extrapolarse a la combinación con otros regímenes de quimioterapia, como lo hicieron dos metaanálisis realizados, añadiendo así consistencia a los resultados anteriores5–7. Asimismo, otros estudios como el pivotal de Slamon et al. demuestran que este beneficio del tratamiento con trastuzumab se hace también extensible a la enfermedad avanzada y en combinación con otros esquemas de quimioterapia y tratamientos hormonales8–11. Por lo tanto el trastuzumab resulta un fármaco fundamental en el manejo de pacientes con cáncer de mama HER2 positivo. Como inconveniente principal cabe resaltar la duración del tratamiento, que en la enfermedad metastásica es hasta progresión o toxicidad inaceptable, lo cual en la mayoría de los casos supone meses de tratamiento (en ocasiones asociados a tratamientos orales: hormonoterapia, quimioterapia oral e incluso otros tratamientos biológicos) y en la situación adyuvante es de un año, tal y como se concluyó de los estudios HERA (un año versus dos) y el estudio PHARE (6 meses versus 1 año)1,2,10,12. Esto supone una carga considerable en los recursos humanos y económicos, tanto para la organización sanitaria como para el propio paciente.
En este contexto, una formulación que permita una administración distinta a la intravenosa tendría importantes beneficios para el paciente y el sistema sanitario, ya que evitaría las constantes visitas al hospital. Para ello se diseñó el desarrollo de una formulación más conveniente, la subcutánea, con el objeto de aunar eficiencia en la utilización de los recursos y facilidad de uso, manteniendo la eficacia.
MétodosEl presente artículo pretende hacer una revisión sistemática de la evidencia actualmente disponible acerca de la formulación subcutánea de trastuzumab, en cuanto a eficacia y seguridad. Para ello se han consultado las bases de datos de PubMed y de ensayos clínicos de ClinicalTrials.gov en busca de ensayos clínicos prospectivos con trastuzumab subcutáneo, fase I, II o III. Las palabras clave utilizadas en esta búsqueda fueron «subcutaneous trastuzumab» y «trial».
Desarrollo de la formulación subcutáneaEl desarrollo de trastuzumab inicialmente se concibió como un esquema de administración semanal, con una dosis de carga de 4mg/kg, seguida de dosis semanales de 2mg/kg. Pero de forma precoz se detectó la necesidad de desarrollar un esquema de administración más favorable a la quimioterapia acompañante. De este modo, un estudio fase II estableció una bioexposición similar entre la pauta semanal y la trisemanal, con parámetros similares de eficacia y tolerancia, a pesar de la esperada mayor concentración pico y la menor concentración de valle del régimen trisemanal13. Este esquema de administración, con una dosis de carga de 8mg/kg administrada en 90 minutos y posteriores dosis de 6mg/kg administradas en 30 minutos, es actualmente la más frecuente para el tratamiento intravenoso acompañando a quimioterapia o a tratamiento endocrinológico.
Teniendo en cuenta los recursos humanos y económicos que requiere la aplicación de una terapia tan altamente eficaz e imprescindible en el tratamiento del cáncer de mama HER2-positivo, cabía esperar el interés que despertaría el desarrollo de una formulación subcutánea.
El hialuronano, un importante componente del tejido extracelular subcutáneo, tiene una vida media en 15-20 horas, y por tanto representa un factor potencialmente modificable en la administración de fármacos por esta vía. La hialuronidasa, enzima encargada de hidrolizar y degradar el hialuronano, se usa desde hace más de 60 años y está aprobada por la Food and Drug Administration de EE. UU. (FDA) en su formulación recombinante, rHuPH20, para facilitar la absorción de otros fármacos coinyectados subcutáneamente. En este estudio, trastuzumab se coinyectaba con rHuPH20 para facilitar la infusión subcutánea de un volumen mayor de lo normalmente admitido en este espacio.
Con este objetivo se diseñó un estudio fase I/Ib que comparaba la administración intravenosa (IV) versus la formulación subcutánea (SC) de trastuzumab y rHuPH20 en voluntarios masculinos sanos y en mujeres diagnosticadas de cáncer de mama HER2 positivo en situación de adyuvancia14. Esto último eliminaba el posible factor de confusión de la influencia del resto tumoral en la farmacocinética.
El objetivo de este estudio fase I/Ib era seleccionar la dosis de trastuzumab subcutáneo que resultaría en una bioexposición similar a la alcanzada con el fármaco intravenoso, mediante el análisis de la curva de concentración sérica-tiempo (AUC). Los objetivos secundarios fueron la tolerancia y los parámetros de seguridad. Los datos farmacocinéticos de la formulación intravenosa en voluntarios sanos y en pacientes resultó similar, lo cual permitía la extrapolación de los datos de la formulación subcutánea de los voluntarios sanos a las pacientes14.
La AUC en las pacientes recibiendo trastuzumab 8mg/kg SC fue comparable con la AUC de las pacientes que recibieron la dosis de 6mg/kg IV. La concentración pico (Cmax) de la formulación subcutánea fue menor debido a su absorción más lenta. Los datos de farmacocinética demostraron que la concentración valle (Ctrough) en el día 22, era comparable con la de la formulación intravenosa a dosis de 6mg/kg. Asimismo, los datos de farmacocinética demostraron la linealidad en un amplio rango de concentraciones séricas, por lo que finalmente 8mg/kg SC fue la dosis elegida para el estudio fase III confirmatorio15.
La biodisponibilidad en voluntarios sanos de las dosis de 6mg/kg, 8mg/kg y 10mg/kg fue de 84%, 91% y 93% respectivamente, mientras que en pacientes esta fue de 87% y 99% respectivamente para las dosis subcutáneas de 8mg/kg y 12mg/kg.
En el estudio fase I, la mayoría de los acontecimientos adversos fueron de índole leve (71% en los voluntarios sanos, y 73% en las pacientes con cáncer de mama), sin eventos graves que reseñar que conllevaran a la suspensión del fármaco, ni a modificaciones en su dosis. La mayoría de los efectos adversos fueron debidos a la inyección local de trastuzumab: eritema y edema en el punto de infusión o molestias en dicha zona. El resto de acontecimientos adversos fueron similares entre ambas formulaciones.
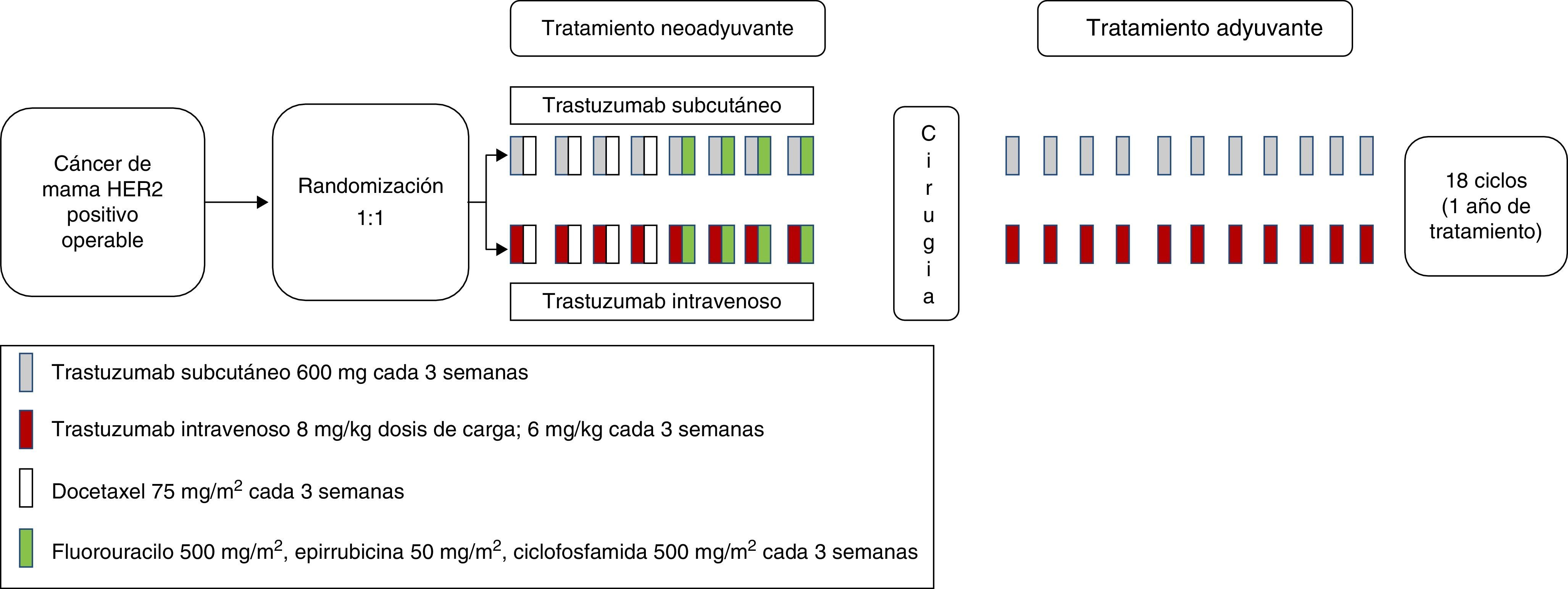
Los datos obtenidos en el estudio fase I/Ib permitieron la realización de un estudio fase III confirmatorio, el estudio HannaH, en pacientes con cáncer de mama HER2-positivos en situación adyuvante o neoadyuvante (fig. 1)15. Este ensayo, randomizado 1:1 y abierto, pretendía establecer equivalencia entre la dosis estándar de trastuzumab intravenoso, y la dosis elegida subcutánea de 600mg, tanto en cuanto a parámetros de farmacocinética (Ctrough previo al ciclo 8, precirugía) como relativos a eficacia (pCR definida como respuesta completa patológica del componente infiltrante en la mama). Los objetivos secundarios fueron el perfil farmacocinético, la respuesta completa patológica tanto en la mama como en la axila (tpCR), la tasa de respuesta, el tiempo a la respuesta, la supervivencia libre de evento, supervivencia global, la seguridad y tolerancia y los datos de inmunogenicidad. Se estipuló que la dosis subcutánea debía alcanzar Ctrough equivalentes o mayores que los obtenidos con la dosis de carga de la formulación intravenosa, 8mg/kg, con la intención de poder omitirla.

Las pacientes en la rama control recibieron trastuzumab neoadyuvante IV acompañado de quimioterapia estándar (4 ciclos de docetaxel 75mg/m2 cada 3 semanas, seguido de 4 ciclos de FEC-75), tras lo cual se realizaba cirugía de resección. Tras ello, continuaban tratamiento adyuvante con trastuzumab intravenoso hasta completar un año. El tratamiento de radioterapia locorregional y tratamiento endocrinológico adyuvante se realizaba a criterio del centro. La rama experimental contemplaba este mismo tratamiento, pero con una dosis de 600mg de trastuzumab SC cada 3 semanas, diluido en 10.000 U rHuPH20 en 5ml. La administración de trastuzumab subcutánea se realizaba mediante jeringa de inyección manual por parte del equipo sanitario.
Se incluyeron un total de 596 pacientes. La intensidad de dosis relativa fue superior a 96% en ambas ramas. Se demostró la no inferioridad de la formulación subcutánea en cuanto a la Ctrough precirugía, con una media geométrica Ctrough de 51,8μg/ml en la rama intravenosa, y 69,0μg/ml en la rama subcutánea. La razón de medias geométricas de Ctrough trastuzumab SC/Ctrough trastuzumab IV fue de 1,33 (IC 90%: 1,24-1,44), mayor que el margen de no inferioridad preespecificado por protocolo, cumpliendo por tanto este objetivo primario. El 98,7% de las pacientes en la rama IV consiguieron un Ctrough mayor de 20μl/ml, versus el 97% de las pacientes en la rama SC, demostrando por tanto la posible omisión de la dosis de carga en la formulación subcutánea.
En cuanto al segundo objetivo primario, pCR en la mama, 45,4% la alcanzaron con la formulación subcutánea, y 40,7% con la intravenosa, con una diferencia de 4,7% (IC 95%: -4,0-13,4%) y por debajo del límite de -12,5% preespecificado para determinar la no inferioridad, cumpliendo por lo tanto también este objetivo. La ocurrencia de efectos adversos fue similar en ambos grupos (52% en ambas ramas). No obstante, se produjeron más efectos adversos graves (grado 3 y 4) en la rama de tratamiento subcutáneo (46,9%) que en el intravenoso (31,2%). Este hecho fue debido principalmente debido a una mayor ocurrencia de infecciones en la rama subcutánea, si bien no hubo diferencias en la toxicidad hematológica que pudiera justificar estas diferencias. No se asoció a más infecciones en el punto de inyección subcutánea, aunque sí que se constató mayor tasa de reacciones en esta zona, principalmente dolor. Se observaron un total de 4 eventos adversos fatales (grado 5), uno en la rama intravenosa y tres en la subcutánea. Aunque 3 de estos cuatro eventos adversos ocurrieron en pacientes obesas o con sobrepeso, el análisis de regresión logística múltiple no pudo arrojar relación alguna entre el peso corporal y las concentraciones séricas de trastuzumab. Sin embargo, resulta difícil asegurar la correcta exposición al fármaco en pacientes obesas dado el escaso número de sujetos con estas características en este estudio.
Por lo tanto, el estudio HannaH demostró equivalencia entre la formulación subcutánea y la intravenosa de trastuzumab en cuanto a parámetros de farmacocinética y eficacia, así como perfil de seguridad y tolerancia. Asimismo, confirmó la posibilidad de obviar la dosis de carga con la formulación subcutánea. Sin embargo, este ensayo no exploró en medida alguna los parámetros de calidad de vida de las pacientes, algo deseable en un estudio que pretendía demostrar una mejora en el tratamiento con trastuzumab mediante una vía de administración más conveniente.
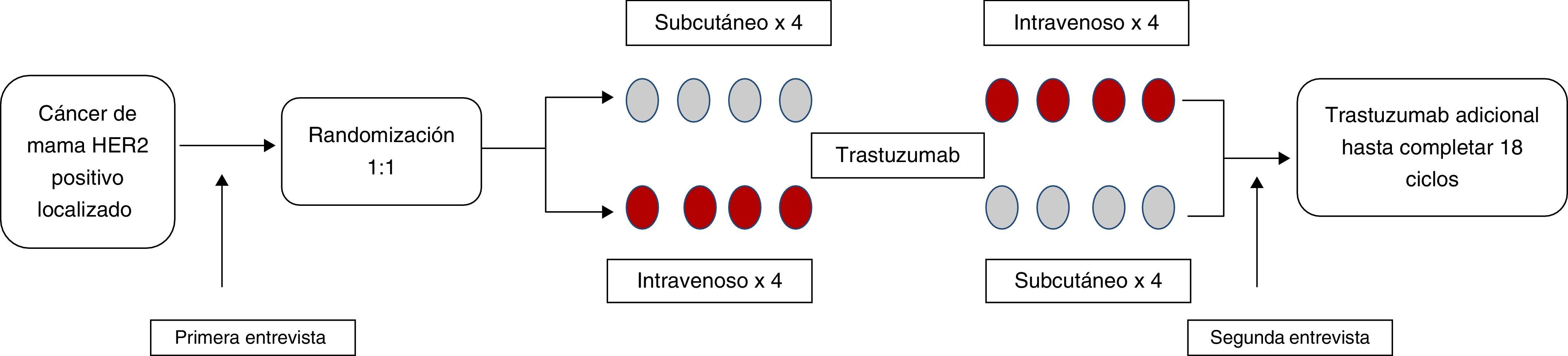
Preferencia del pacienteSe han diseñado varios estudios para determinar la preferencia de las pacientes entre la formulación subcutánea y la clásica intravenosa. El más significativo hasta la fecha, el estudio PrefHer, en situación adyuvante, contemplaba una primera cohorte (A) de 124 pacientes en la que recibían trastuzumab SC 600mg/5ml cada 3 semanas durante 4 ciclos, administrado por un profesional sanitario en el centro habitual donde recibían el resto del tratamiento, tras lo cual cambiaban a recibir la formulación intravenosa (fig. 2)16. En la segunda cohorte (B), de 124 pacientes, estas recibían primero 4 ciclos de formulación intravenosa, seguido de trastuzumab subcutáneo. Ambas ramas completaban un año de tratamiento adyuvante. El objetivo primario del estudio era determinar la proporción de pacientes con preferencia por la vía subcutánea o la intravenosa. Se realizaban dos entrevistas telefónicas, una antes de la randomización, y otra tras 8 ciclos de tratamiento (es decir, tras completar 4 ciclos de la primera formulación y 4 de la alternativa).

La mayoría de los efectos adversos en el estudio PrefHer, recogidos en el periodo de cruce de tratamientos, fueron de grado 1 o 2. Se objetivaron ligeramente más efectos adversos en el periodo de administración de la formulación subcutánea, principalmente a expensas de reacciones cutáneas en el punto de inyección (8% versus 0%), así como reacciones relacionadas con la administración (9% versus 5%). No se objetivaron diferencias significativas en la cardiotoxicidad entre ambas formulaciones. La tasa de descontinuación de trastuzumab fue baja en ambas ramas (0,8% versus 0,4%).
Finalmente, 117 pacientes en la cohorte A y 119 pacientes en la cohorte B fueron incluidas en el análisis por intención de tratar. El 91,5% de las pacientes mostraron preferencia por la vía subcutánea (IC 95%: 87,2-94,7% p<0,0001), 6,8% prefirieron la intravenosa (IC 95%: 3,9-10,8%) y 1,7% no mostraron ninguna preferencia. Este estudio también analizó como objetivo secundario la preferencia de los profesionales sanitarios: 73,8% prefirieron la administración subcutánea (IC 95%: 64,2-82,0%) sobre la intravenosa (1,9%) y 24,3% no mostraron ninguna preferencia.
Existen dos estudios en marcha que evalúan aspectos de preferencia y calidad de vida de la formulación subcutánea. El estudio SafeHer de preferencia tiene previsto incluir aproximadamente 2.500 pacientes con cáncer de mama en estadios precoces, y su objetivo es el análisis de tolerancia y seguridad de la formulación subcutánea de uso único y la jeringa manual. Asimismo, el estudio ChangHer, también en marcha, pretende explorar estos mismos parámetros entre la formulación subcutánea mediante el dispositivo autoinyectable de uso único o la formulación intravenosa en población metastásica.
Adicionalmente, otros estudios ya en reclutamiento o recientemente terminados compararán las diferentes vías de administración de trastuzumab subcutáneo y ahondarán en los parámetros de calidad de vida de los pacientes (estudios HOMERUS, LISAH, SCHEARLY, HerSCin, NCT01964391 y NCT01926886), lo cual contribuirá a definir los potenciales beneficios de esta vía de administración. Asimismo, estos estudios, algunos de ellos de uso expandido contribuirán a definir la eficacia de trastuzumab en pacientes infrecuentes en los estudios precedentes, como las pacientes con índices de masa corporal extremos.
Análisis de coste-eficaciaUn análisis de coste eficacia realizado en paralelo al estudio PrefHer en el Reino Unido (Time & Motion) evaluó el coste eficacia de la administración subcutánea de trastuzumab mediante dispositivo autoinyectable de único uso comparado con la intravenosa17. La primera suponía un ahorro de 2.440 € por cada año de administración y paciente con cáncer de mama en estadio precoz, principalmente debido a menores costes en tiempo de administración, aunque también de preparación y consumibles necesarios para la misma. En este estudio solo se analizaron los costes derivados de la administración, y no aquellos secundarios a toxicidad. Sin embargo, los datos derivados del estudio fase I y del fase III (estudio HannaH) no mostraron una mayor tasa de efectos secundarios, por lo que no cabe esperar un incremento en el coste derivado de estos.
Asimismo, otro estudio observacional prospectivo perteneciente a Time & Motion similar al anteriormente descrito, confirmó los resultados anteriores, mostrando una reducción en tiempo de administración de entre 58-81% según país, así como en el tiempo de preparación farmacéutica y tiempo sanitario18.
ConclusionesTrastuzumab ha supuesto un cambio radical en el tratamiento de las pacientes con cáncer de mama HER2 positivo, tanto en situación precoz, (neo)-adyuvancia, como en la enfermedad metastásica. Aproximadamente el 15% de los tumores de mama son HER2 positivos. Sin embargo, el tratamiento prolongado establecido en todas las situaciones, y la necesidad hasta el momento de una administración intravenosa periódica, ha supuesto una gran carga asistencial en los hospitales de día oncológicos y en los servicios de farmacia.
La formulación subcutánea de trastuzumab con una dosis fija de 600mg, recientemente aprobada por las autoridades sanitarias, supone un gran avance en este sentido. Teniendo en cuenta el mejor perfil de coste-eficacia, junto con la preferencia de las pacientes y la facilidad de administración, la formulación subcutánea se perfila como favorable de cara a su combinación con esquemas de quimioterapia o tratamientos hormonales orales. Además su potencial administración en centros de salud ambulatorios o por las propias pacientes permitiría dotar de una mayor independencia del ámbito hospitalario. Se evitarían los problemas asociados a los dispositivos de infusión intravenosa de larga duración siendo además previsible que el dispositivo autoinyectable de único uso suponga una mayor reducción en el coste, y mayor comodidad para las pacientes. Los estudios de farmacocinética y farmacodinámica confirman una equivalencia entre la formulación subcutánea y la intravenosa, sin suponer mayor toxicidad.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

