



La familia «Toll-like» constituye un importante grupo de receptores de reconocimiento de patrones cuyos ligandos incluyen una amplia variedad de moléculas con una fuerte actividad adyuvante (lipopolisacárido, lipopéptidos y DNA bacteriano). Estos ligandos pueden activar células dendríticas, macrófagos y otras células presentadoras que mostrarán antígenos microbianos a células del sistema inmune adaptativo. Actualmente, la identificación y caracterización de ligandos endógenos para estos receptores ha proporcionado una nueva perspectiva para el estudio de la etiología de algunas enfemedades autoinmunes. En lugar de considerar la autoinmunidad como una respuesta aberrante hacia antígenos del propio organismo por parte del sistema inmune adaptativo, al menos en algunos casos, podemos también decir que el fenómeno autoinmune viene de una respuesta hacia ligandos exógenos o endógenos por parte del sistema inmune innato. En esta revisión se analizan datos recientemente publicados que señalan una importante conexión entre inmunocomplejos conteniendo DNA y RNA, la activación de receptores «Toll-like», la producción de interferones de tipo I (INF-α, INF-β) y el desarrollo de algunas enfermedades sistémicas autoinmunes.
The Toll-like receptor family is an important group of pattern-recognition receptors whose ligands include a wide range of molecules with strong adjuvant activity (such as lipopolysaccharide, lipopeptides and bacterial DNA). These ligands can activate dendritic cells, macrophages and other antigen presenting cells that allow the effective presentation of microbial antigens to cells of the adaptive immune system. Nowadays, the identification and characterization of endogenous ligands for these receptors has provided a novel perspective for examining the etiology of some autoimmune diseases. Instead of being considered as an aberrant response to host antigens by the adaptive immune system, autoimmunity can be viewed as arising from a response to exogenous or endogenous ligands by the innate immune system, at least in some cases. This review summarizes recently published data that indicate an important connection between DNA-and RNA-containing immune complexes, activation of Toll-like receptors, production of type I interferons (INF-α, INF-β) and the development of some systemic autoimmune diseases.
Una de las propiedades principales del sistema inmune en mamíferos es la habilidad de generar una respuesta efectiva frente a diferentes patógenos, manteniendo al mismo tiempo un nivel de tolerancia hacia los propios tejidos. Durante la pasada década y hasta la actualidad, el conocimiento de los componentes moleculares y de la función de la inmunidad innata en la defensa del huésped ha ido creciendo de manera vertiginosa. Desde el primer momento se ha visto que el reconocimiento de la mayoría de los microorganismos está mediado por varias familias de receptores que colectivamente «vigilan» el espacio extracelular, los compartimentos endosomales y el citoplasma, siendo sensibles a la detección del mínimo signo de infección. Estos Receptores de Reconocimiento de Patrones («PRR»), que son codificados por genes en la línea germinal, reconocen Patrones Moleculares Asociados a Patógenos («PAMP») y activan células de la rama innata del sistema inmune, incluyendo células dendríticas (CD)1,2. Actualmente existen múltiples evidencias de que los PRR no solo reconocen estructuras de patógenos sino que también están involucrados en la respuesta al daño tisular y aclaramiento de cuerpos apoptóticos3. Dentro de este grupo de receptores de respuesta innata se incluyen los TLR (Toll-like receptors), RLR (RIG-I, retinoic acid-inducible gene-I,-like receptors) y NLR (Nod-like receptors)4.
La familia de los receptores «Toll-like» constituye uno de los grupos más importantes dentro de los PRR. Los ligandos microbianos que reconocen incluyen una amplia variedad de moléculas con fuerte actividad adyuvante (lipopolisacárido, lipopéptidos y DNA bacteriano, entre otras) que pueden activar células dendríticas, macrófagos y otras células presentadoras de antígenos permitiendo una presentación efectiva a células del sistema inmune adaptativo. También han sido identificadas, un cierto número de moléculas endógenas (DNA, RNA, etc.) que actúan como ligandos de algunos TLR3.
En las enfermedades autoinmunes existe una alta proporción de autoanticuerpos que unen DNA, RNA o complejos macromoleculares que contienen estas 2 moléculas (Lupus Sistémico Eritematoso [LES], Esclerodermia y síndrome de Sjögren). El por qué estos epítopos han tenido un papel tan importante en autoinmunidad ha sido una cuestión muy debatida. Una posibilidad es que estas moléculas se acumulen en la membrana plasmática durante la muerte celular por apoptosis5, siendo su posterior procesamiento por parte de células presentadoras de antígeno, el que conduciría a la pérdida de tolerancia frente a las mismas. Otra posibilidad es que se creen neo-epítopos que sean reconocidos por células del sistema inmune adaptativo tras el procesamiento de estas moléculas por granzima B u otras proteasas/nucleasas, o tras modificaciones postraduccionales6,7. Además, persiste la idea de que estos autoantígenos pueden actuar como «autoadyuvantes», es decir, tienen la capacidad de activar el sistema inmune innato directamente, a través de receptores como los TLR que reconocen, entre otros, ligandos endógenos3.
En esta revisión se pretenden analizar e integrar los datos publicados recientemente relacionados con la estructura, reconocimiento y señalización por parte de los TLR, así como la sucesión de eventos que pueden conectar el desarrollo y/o la progresión de enfermedades autoinmunes sistémicas con la función de estos receptores.
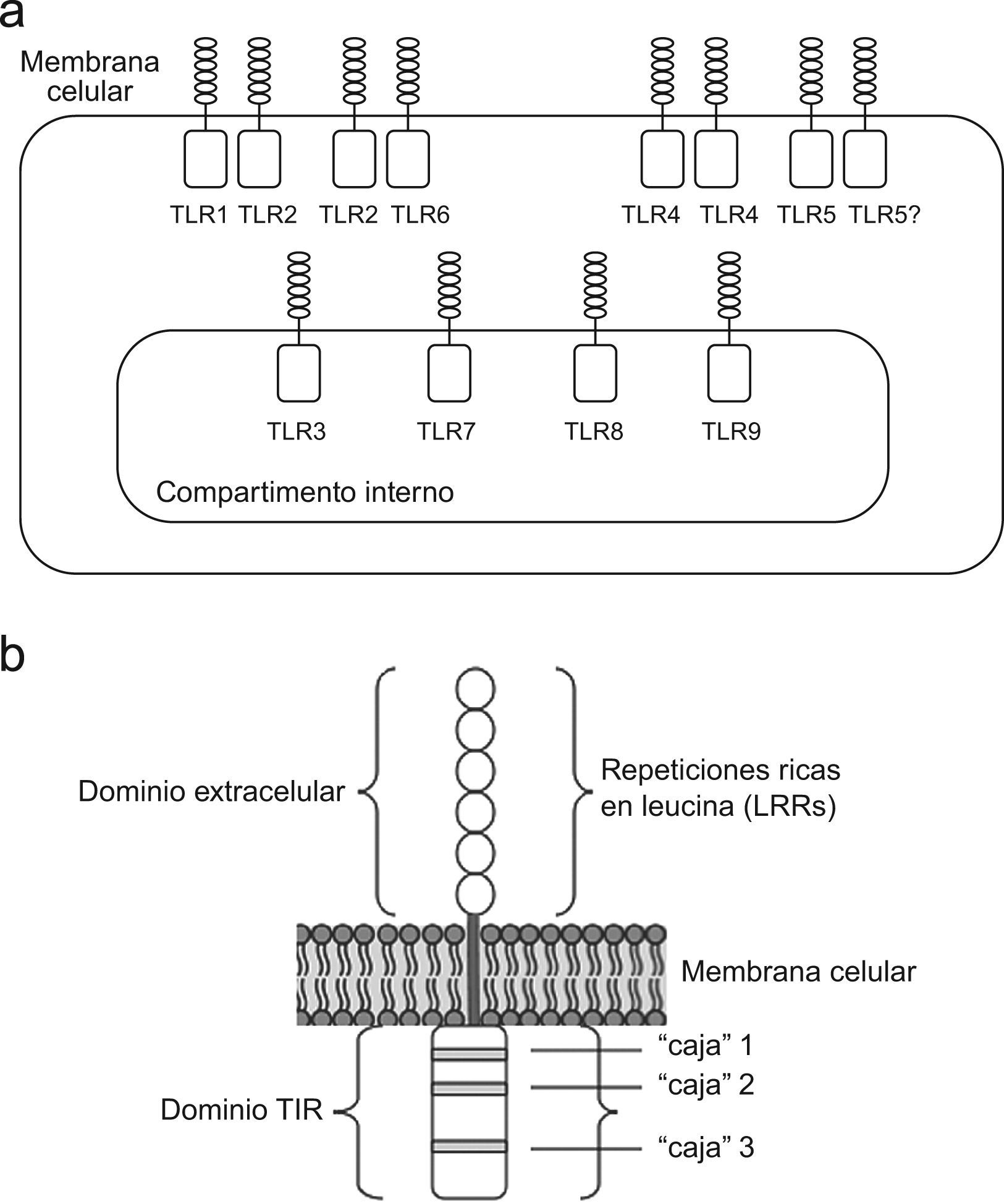
TLR: estructura y reconocimiento de ligandosLos receptores «Toll-like» representan una familia de PRR conservados evolutivamente, que reconocen una amplia variedad de componentes microbianos8–12. En mamíferos se han descrito 11 tipos diferentes, encontrándose la mayoría de ellos ampliamente distribuidos en diferentes tipos celulares del sistema inmune, incluyendo células dendríticas, macrófagos, células «natural killer» (NK), mastocitos, neutrófilos y linfocitos T y B. Se encuentran además en células no relacionadas con el sistema inmune como son los fibroblastos, células epiteliales y queratinocitos13. La mayoría de estos receptores se expresan en la superficie celular (TLR1, 2, 4, 5, 6, 10 y 11) mientras que una minoría se encuentran presentes en compartimentos endosomales (TLR3, 7, 8 y 9) (fig. 1a). Los TLR se pueden expresar como homodímeros o como heterodímeros (TLR2+TLR1 o TLR2+TLR6).
 Localización celular de los receptores «Toll-like» (Kuby 6a edición, modificado). b) Estructura de un receptor «Toll-like» (Kuby 6.a edición, modificado).")
En relación a su estructura, son proteínas de membrana que comparten un elemento común en su región extracelular, repeticiones de segmentos de 24–29 aminoácidos que contienen la secuencia xLxxLxLxx, repeticiones ricas en leucina, LRR (x, cualquier aminoácido, L es leucina) (fig. 1b). El dominio intracelular se denomina dominio TIR (de Toll/receptor IL-1) debido a la similitud entre los dominios citoplásmicos de los TLR y del receptor de la IL-1. Estos dominios presentan 3 regiones altamente conservadas entre todos los miembros de la familia TIR, denominadas «cajas» 1, 2 y 3, que son los lugares de unión para proteínas intracelulares que participan en las vías de señalización mediadas por TLR14.
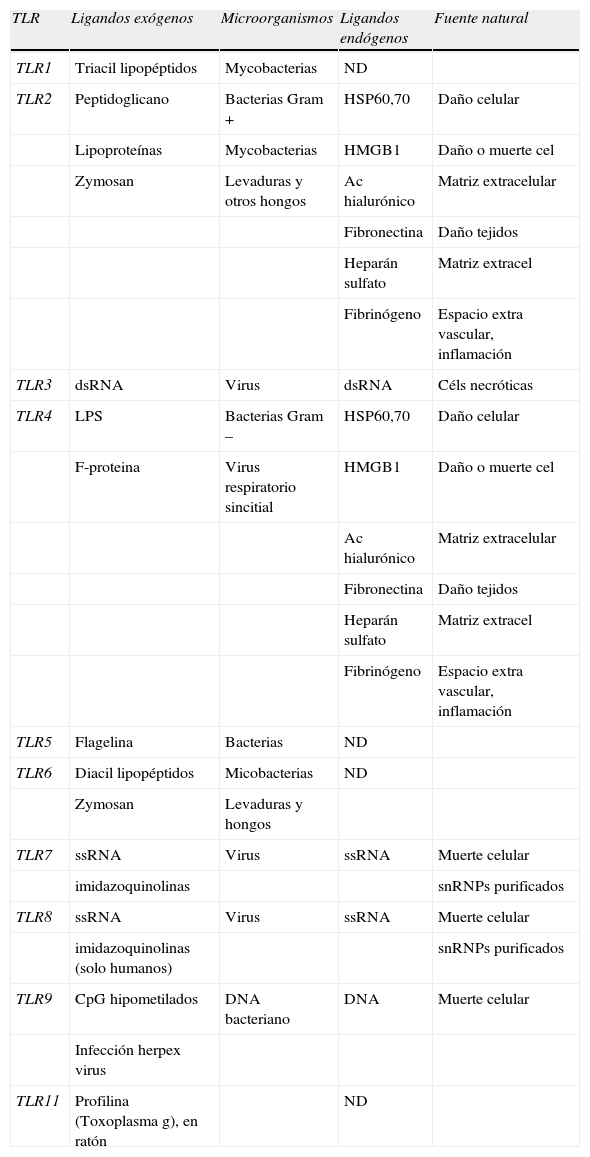
Recientemente algunos de los TLR descritos han sido cristalizados (TLR-1-TLR-2, TLR-3 y TLR-4). Se piensa que las estructuras obtenidas son responsables de la unión de ligandos y de la dimerización inducida en algunos de ellos para el reconocimiento15–18. Cada TLR reconoce patrones específicos de componentes microbianos (tabla 1).
Ligandos endógenos y exógenos de los receptores Toll-like
| TLR | Ligandos exógenos | Microorganismos | Ligandos endógenos | Fuente natural |
| TLR1 | Triacil lipopéptidos | Mycobacterias | ND | |
| TLR2 | Peptidoglicano | Bacterias Gram + | HSP60,70 | Daño celular |
| Lipoproteínas | Mycobacterias | HMGB1 | Daño o muerte cel | |
| Zymosan | Levaduras y otros hongos | Ac hialurónico | Matriz extracelular | |
| Fibronectina | Daño tejidos | |||
| Heparán sulfato | Matriz extracel | |||
| Fibrinógeno | Espacio extra vascular, inflamación | |||
| TLR3 | dsRNA | Virus | dsRNA | Céls necróticas |
| TLR4 | LPS | Bacterias Gram – | HSP60,70 | Daño celular |
| F-proteina | Virus respiratorio sincitial | HMGB1 | Daño o muerte cel | |
| Ac hialurónico | Matriz extracelular | |||
| Fibronectina | Daño tejidos | |||
| Heparán sulfato | Matriz extracel | |||
| Fibrinógeno | Espacio extra vascular, inflamación | |||
| TLR5 | Flagelina | Bacterias | ND | |
| TLR6 | Diacil lipopéptidos | Micobacterias | ND | |
| Zymosan | Levaduras y hongos | |||
| TLR7 | ssRNA | Virus | ssRNA | Muerte celular |
| imidazoquinolinas | snRNPs purificados | |||
| TLR8 | ssRNA | Virus | ssRNA | Muerte celular |
| imidazoquinolinas (solo humanos) | snRNPs purificados | |||
| TLR9 | CpG hipometilados | DNA bacteriano | DNA | Muerte celular |
| Infección herpex virus | ||||
| TLR11 | Profilina (Toxoplasma g), en ratón | ND |
ND: no determinado.
TLR1: se encuentra funcionalmente asociado a TLR219. Análisis de ratones deficientes en TLR1 han demostrado la importancia de este receptor en el reconocimiento de triacil-lipopéptidos20. Por otra parte, macrófagos de ratones «knockout» para TLR1 muestran una reducción en la producción de citoquinas inflamatorias en respuesta a triacil-lipopéptidos y lipoproteínas de micobacterias21.
TLR2: reconoce un amplio rango de PAMP que incluyen lipoproteínas de bacterias Gram-negativas, Mycoplasma fermentans, Treponema pallium y Borrelia burgdorferi, ácido lipoteicoico y peptidoglicano de bacterias Gram-positivas, lipoarabinomanano de micobacterias, etc22. El mecanismo por el que TLR2 reconoce una amplia variedad de componentes microbianos se explica por el hecho de que forma dímeros con TLR1 y TLR6, como se ha comentado anteriormente.
TLR3: reconoce RNA de doble cadena (dsRNA) que es sintetizado por la mayoría de los virus durante su replicación23, detectando por tanto infecciones virales. El dsRNA induce la síntesis de interferones de tipo I (INF-α/β) que ejercen un efecto antiviral e inmunoestimulatorio a través de la maduración de células dendríticas24. TLR3 se expresa en los endosomas de células inmunes, incluyendo células dendríticas convencionales (cDC), macrófagos, linfocitos B, células NK y células no relacionadas con el sistema inmune, como son las células epiteliales25. Sin embargo, no se expresa en células dendríticas plasmacitoides (pDC) que producen grandes cantidades de interferones de tipo I. A pesar de todos estos datos, no está clara la función de TLR3 en las respuestas antivirales debido a que existen trabajos que ponen en evidencia que ratones «knockout» para TLR3 no muestran más susceptibilidad a las infecciones por ciertos virus que sus correspondientes análogos «wild-type»26.
TLR4: Es esencial para el reconocimiento del lipopolisacárido bacteriano (LPS), componente mayoritario de la membrana externa de las bacterias Gram-negativas, con una potente actividad inmunoestimulatoria27,28. Para este reconocimiento requiere moléculas adicionales como CD14, expresado en monocitos/macrófagos y neutrófilos. TLR4 directa o indirectamente también reconoce moléculas endógenas como proteínas de choque térmico («heat shock proteins»)29, fibrinógeno, ácido hialurónico y β-defensina, entre otras30,31. Así esta molécula parece estar implicada en el reconocimiento de ligandos endógenos relacionados con la respuesta inflamatoria, independientemente de la fuente de infección.
TLR5: reconoce un monómero de flagelina, proteína estructural de los flagelos de las bacterias. Este receptor se expresa en la membrana basolateral de las células epiteliales intestinales y en las superficies luminales de las células que recubren la tráquea, bronquios y alveolos, lo que indica el papel tan importante que tiene en el reconocimiento microbiano a nivel de las superficies de las mucosas.
TLR6: como en el caso de TLR1, TLR6 se encuentra asociado funcionalmente con TLR2. La coexpresión de ambos receptores es un requisito indispensable para el reconocimiento de diacil lipopéptidos derivados de micoplasma32,33.
TLR7: se expresa en las membranas endosomales y en un principio se ha visto que está involucrado en la respuesta inmune a las imidazoquinolinas, usadas en el tratamiento de infecciones virales. Posteriormente se ha observado que puede reconocer RNA de cadena sencilla (ssRNA) procedente de virus como el VIH, virus de la estomatitis vesicular y virus influenza34,35. El ssRNA también se puede producir en el huésped, pero no es reconocido por TLR7 porque en condiciones normales, esta molécula no accede a estos compartimentos celulares. Sin embargo, cuando el ssRNA se conjuga con un anticuerpo anti-ssRNA, este inmunocomplejo se libera dentro del endosoma de las pDC mediante endocitosis, es reconocido por TLR7, con la consiguiente producción de INF-α36. Además se ha visto que el ssRNA se puede unir de manera ocasional al receptor del linfocito B (BCR) de células autorreactivas, que puede conducir a la liberación de ssRNA en el endosoma, por tanto induciendo la producción de autoanticuerpos37,38.
TLR8: el gen que codifica para este receptor presenta mucha homología con el gen del TLR7, ambos localizados en el cromosoma X. En humanos se ha comprobado que TLR8 reconoce las imidazoquinolinas y ssRNA, que son los ligandos del TLR7. Como característica distintiva, se ha observado que TLR8 se expresa en células reguladoras (Treg) y su activación produce una cierta inhibición de la función de las mismas39.
TLR9: es esencial para el reconocimiento de motivos no metilados CpG de DNA procedente de bacterias y virus. Hay al menos 2 tipos de motivos CpG descritos, A y B, ambos reconocidos por TLR9. CpGB, es el motivo convencional, que fue originariamente identificado como un potente inductor de citoquinas inflamatorias como IL-6, IL-12 y TNF-α y regula positivamente moléculas coestimulatorias como CD80, CD86 y MHC cl II en células pDC y en menor extensión linfocitos B40. CpGA es estructuralmente diferente y presenta una gran habilidad para inducir la producción de INF-α por parte de las pDC, y menor habilidad para inducir IL-1241.
De forma similar a TLR7, TLR9 está involucrado en la patogénesis de enfermedades autoinmunes3. La producción de factor reumatoide por linfocitos B autorreactivos está mediada por la unión del inmunocomplejo IgG2a-cromatina por parte del BCR y TLR942.
TLR10: este receptor se ha identificado como una molécula estrechamente relacionada a TLR1 y TLR6 aunque su ligando por el momento es desconocido.
TLR11: es una molécula no funcional en humanos, ya que presenta un codón stop en el gen TLR11.
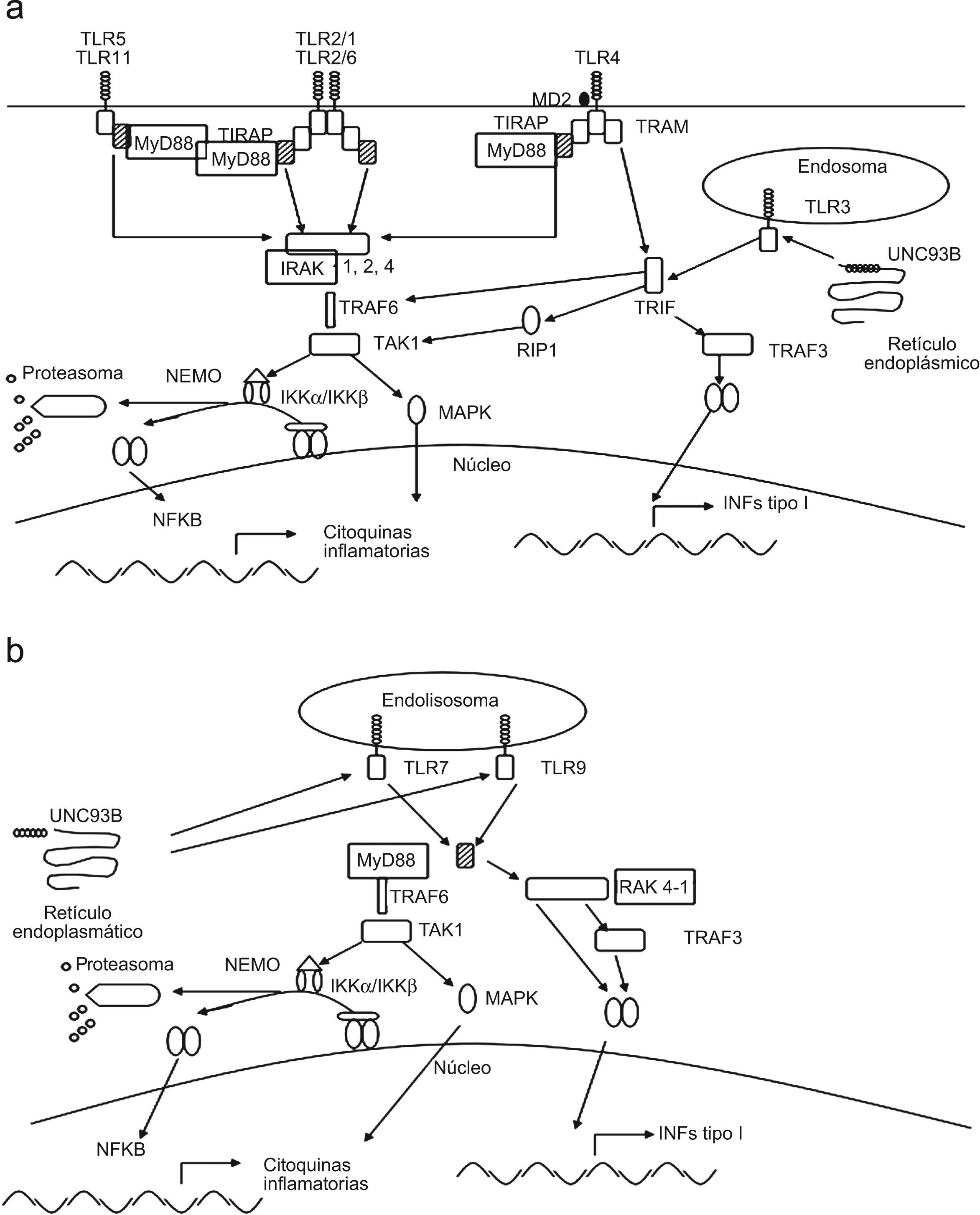
Vías de señalización de los TLREl reconocimiento de agentes microbianos por parte de los TLR facilita en muchos casos la heterodimerización de los mismos, TLR2 dimeriza con TLR1 o TLR6, en otros casos sin embargo, estos receptores pueden homodimerizar (dímeros de la misma molécula). En ambas situaciones, la dimerización de los TLR activa la cascada de señalización que se origina en el dominio TIR (fig. 1b). Existe un adaptador denominado MyD88 común a la señalización de todos los TLR, excepto TLR3, que es imprescindible para la inducción de citoquinas inflamatorias, TNF-α e IL-12. Estudios recientes han puesto en evidencia que las vías de señalización a través de los TLR pueden ser divergentes, detectándose una vía dependiente de MyD88 y otra independiente43,44 (fig. 2a y b).
TLR y enfermedadInmunodeficiencias Señalización vía receptores «Toll-like» en células dendríticas convencionales o macrófagos. b) Señalización vía receptores «Toll-like» en células dendríticas plasmacitoides.")
Variaciones genéticas o mutaciones detectadas en genes que codifiquen TLR y algunas de las proteínas de la cascada de señalización, se han visto implicadas en la predisposición a desarrollar cuadros infecciosos relacionados con deficiencias de la rama innata de la inmunidad45. Una de las publicaciones más recientes en este ámbito ha sido la descripción de la deficiencia autosómica recesiva del adaptador «MyD88» en pacientes pediátricos. Una de las características más importantes de la clínica que presentan estos pacientes, es la susceptibilidad a las infecciones por bacterias pirógenas como Streptococcus pneumoniae y Staphylococcus aureus. La severidad de la clínica mejora con la edad y se ha visto que no son susceptibles a otro tipo de microorganismos. Estas observaciones han sugerido que la vía de señalización dependiente de MyD88 es esencial para la protección inmune frente a unas pocas bacterias, sin embargo resulta redundante para la defensa del huésped frente a la mayoría de las infecciones46.
También se ha descrito la deficiencia asociada a «IRAK4» (quinasa 4 asociada al receptor de la IL-1) (fig. 2a y b) en 28 pacientes que muestran susceptibilidad a infecciones por bacterias Gram-positivas y Gram-negativas cuyas manifestaciones clínicas aparecen antes de la adolescencia y como en el caso anterior, mejoran con la edad. Estos pacientes también muestran una disminución en la producción de interferones de tipo I, aunque no presentan susceptibilidad a infecciones virales, parásitos u hongos47.
Pacientes que muestran una susceptibilidad incrementada hacia la infección por virus herpex (HSV-1) se ha visto que presentan una deficiencia de «TLR3»48. La deficiencia de «UNC93B» también ha sido documentada en pacientes con encefalitis por virus herpex simplex, estos enfermos no responden a TLR3, TLR7, TLR8 o TLR949.
Un defecto clave en la Inmunodeficiencia Común Variable (CVID) es probablemente la pérdida de función TLR7 y TLR9 debido a que la activación de estos receptores parece ser un mecanismo importante en la supervivencia de las células B memoria50. Trabajos recientes han puesto en evidencia que defectos de TLR9 en los linfocitos B, hacen que estas células no sean activadas por CpG incluso habiendo coestimulación mediante el BCR51. Por otra parte, la estimulación de linfocitos B CD27+ y CD27- de estos pacientes con agonistas para TLR7/8, no produce proliferación mediada por TLR, cambio de isotipo y producción de inmunoglobulinas52.
Con todos estos datos podemos observar que las evidencias van aumentando a favor de que los TLR juegan un papel muy importante en la patogénesis de algunas inmunodeficiencias, hasta ahora de origen desconocido.
AutoinmunidadExiste una alta proporción de autoanticuerpos que se unen bien a DNA, RNA o a complejos que contienen estas moléculas y que están relacionados con enfermedades autoinmunes sistémicas (Lupus Eritematoso Sistémico, LES, Esclerodermia, síndrome de Sjögren). Ya se ha comentado anteriormente que estos epítopos pueden hacerse visibles por acúmulo en las membranas, en el caso de muerte celular o aparecer como epítopos de nueva creación, cuando surgen a consecuencia del procesamiento de estas moléculas por proteasas o nucleasas.
En los siguientes apartados se analizará la importante conexión que existe entre la activación de células dendríticas vía TLR, la formación de inmunocomplejos con DNA y RNA y la producción de interferones de tipo I (INF-α/β) por parte de estas células. Esta sucesión de eventos se ha relacionado, en algunas ocasiones, con el desarrollo y/o la progresión de algunas enfermedades autoinmunes sistémicas.
- a)
Subpoblaciones de células dendríticas:
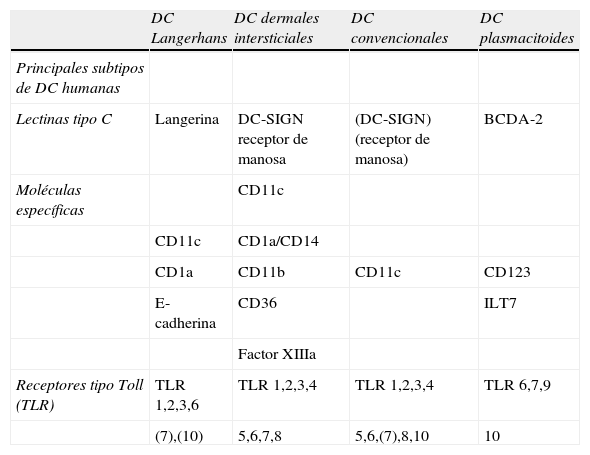
Actualmente las DC constituyen un sistema integrado de células presentadoras que está compuesto por distintos subtipos celulares que expresan diferentes receptores microbianos53, distintas moléculas de superficie54,55, así como un perfil de citocinas propio (tabla 2). In vivo pueden identificarse varios tipos de células dendríticas en base a sus progenitores, distribución tisular y a los marcadores que expresan en superficie56–58. Sin embargo, en la mayoría de los trabajos realizados sobre los subtipos de DC humanos su definición se ha restringido principalmente a las subpoblaciones detectadas en sangre periférica. En el hombre, aproximadamente el 0,1% de las células mononucleares de sangre periférica son DC.
Tabla 2.Principales subtipos de células dendríticas humanas (cedida por la Dra. Mar Naranjo Gómez)
DC Langerhans DC dermales intersticiales DC convencionales DC plasmacitoides Principales subtipos de DC humanas Lectinas tipo C Langerina DC-SIGN receptor de manosa (DC-SIGN) (receptor de manosa) BCDA-2 Moléculas específicas CD11c CD11c CD1a/CD14 CD1a CD11b CD11c CD123 E-cadherina CD36 ILT7 Factor XIIIa Receptores tipo Toll (TLR) TLR 1,2,3,6 TLR 1,2,3,4 TLR 1,2,3,4 TLR 6,7,9 (7),(10) 5,6,7,8 5,6,(7),8,10 10 En base a la expresión del marcador mieloide CD11c se han identificado 2 subtipos mayoritarios de DC conocidos como células dendríticas convencionales o mieloides CD11c+ (cDC o mDC) y las células dendríticas plasmacitoides CD11c- (pDC)53–55. Ambos subtipos se encuentran tanto en sangre periférica de adulto como en sangre de cordón umbilical59 y presentan un patrón diferencial de citocinas60 y de receptores de quimiocinas61, una distinta localización microambiental, un diferente potencial de migración62 así como también un perfil diferenciador en la expresión de otros marcadores de superficie entre los que destacan los PRR63.
- b)
Células dendríticas y su especialización funcional:
Las DC son las células del sistema inmune innato con mayor capacidad activadora y moduladora del sistema inmune adaptativo56. Estas células se encuentran ampliamente distribuidas en los tejidos de distintos órganos64,56, particularmente en aquellas zonas de interfase con el medio externo (piel y mucosas)56, representando el 1–2% del total de células. Este tipo celular está equipado con una gran variedad de PRR, incluyendo TLR65, receptores que como se ha comentado anteriormente, se expresan en la superficie celular, donde reconocen bacterias y hongos, o también en los compartimentos endosomales, donde detectan ácidos nucleicos procedentes de virus. La activación de estos receptores produce activación y maduración de DC43 que favorece la expresión de moléculas MHC de clase II (señal 1) y moléculas coestimuladoras como CD80 y CD86 (señal 2). Además, la activación mediada por los TLR induce la expresión de varias proteínas de membrana y solubles, que son críticas para la inducción de la mayoría de las funciones efectoras de los linfocitos T CD4+ y CD8+ (señal 3).
Dependiendo del tipo de célula dendrítica y de la señal transmitida a las células T CD4 «naïve», estas se pueden diferenciar en distintos subtipos Th, que se caracterizan por diferentes perfiles de citoquinas y funciones efectoras tal y como se describió inicialmente66. Th1 y Th17, fenotipo recientemente identificado, producen citoquinas proinflamatorias INF-γ e IL-17, respectivamente67. Las células Th1 son cruciales para la inducción de las respuestas de linfocitos B y T citotóxicos. Además, Th1 y Th17 se ha visto que son mediadores críticos en enfermedades autoinmunes mediadas por linfocitos T68. El fenotipo Th2, que principalmente secreta IL-4, es responsable de la inducción de respuestas humorales y del aclaramiento de patógenos extracelulares y puede ejercer un efecto protector en ciertas enfermedades autoinmunes69.
Las pDC producen INF-α en respuesta a una infección viral y por tanto tienen un papel muy importante en la activación de la rama innata del sistema inmune.
Por otra parte, se ha comprobado que inmunocomplejos aislados de pacientes con LES, pueden ser potentes estimuladores para la producción de INF-α especialmente por poblaciones de pDC que han sido previamente expuestas a interferones tipo I u otras citoquinas como el factor estimulante de colonias granulocitos/macrófagos (GMCSF)70. Además, las pDC pueden activarse por IgGs derivadas de pacientes, mezcladas con restos celulares provenientes de necrosis o apoptosis, debido a que expresan receptores para el Fc de la IgG (FcγR), que unen e internalizan estos complejos actuando como receptores activadores71,72.
- c)
Las pDC expresan TLR que detectan inmunocomplejos con ácidos nucleicos:
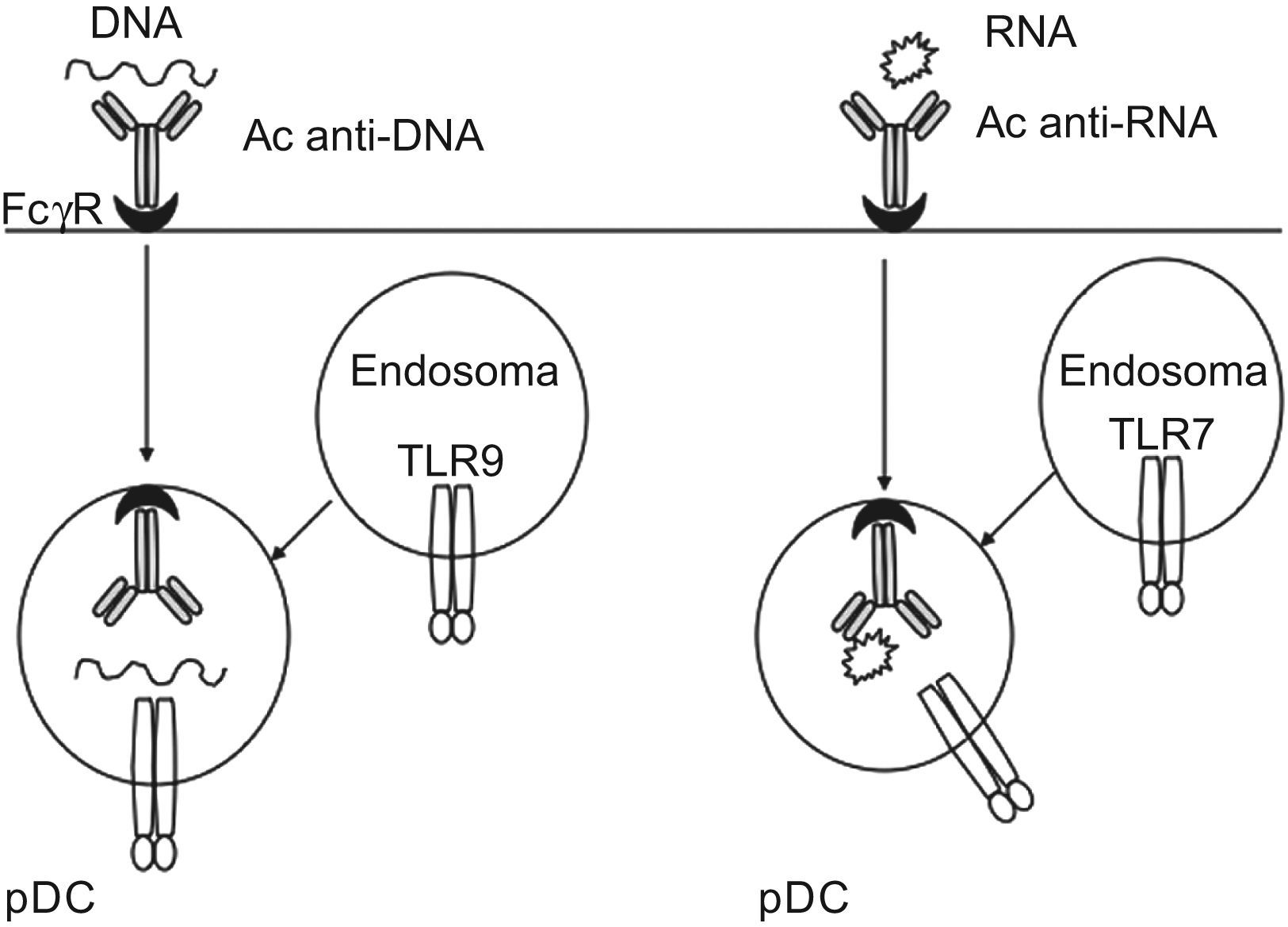
Una de las razones por las que las pDC responden de manera muy eficiente a las infecciones microbianas y a los inmunocomplejos es que además de otros PRR, estas células constitutivamente expresan TLR7 y TLR963. Como se ha comentado previamente, TLR9 fue originalmente identificado como un receptor que podía distinguir entre DNA bacteriano, o viral, con motivos CpG hipometilados del DNA perteneciente a mamíferos; TLR7 se identificó como receptor de ssRNA viral.
La conexión que se produce entre los inmunocomplejos, los receptores Fc de las inmunoglobulinas (FcγR) y el TLR9 ha sido establecida por diferentes grupos de investigadores73,74. El compartimento celular exacto en el que estos componentes interactúan es probablemente dependiente del tipo celular, aunque se ha visto que los endosomas son muy relevantes en las pDCs75.
De manera similar a lo observado con TLR9, los inmunocomplejos que contienen RNA activan diferentes tipos celulares a través de TLR7. Los sueros de pacientes con LES o síndrome de Sjögren, a menudo tiene elevados títulos de anticuerpos específicos para pequeñas ribonucleoproteínas nucleares (snRNP), que son complejos macromoleculares formados por RNA asociado a proteínas. Aunque ha sido muy complicado mostrar que esos sueros activen pDC directamente, se ha confirmado que IgG purificada de pacientes con LES induce la producción de INF-α, si se mezcla con snRNP purificados. Esta producción de INF-α por parte de las pDC, se inhibe con cloroquina o bafilomicina, agentes que interfieren con la acidificación de los endosomas y bloquean la activación de TLR7 y TLR9. La producción de citoquinas también se bloquea utilizando secuencias de oligodeoxinucleótido (ODN) que inhiben la activación de los TLR7 y TLR976,36. Todos estos estudios realizados in vitro, fuertemente sugieren que la unión de estos inmunocomplejos, mediada por FcγR, a los TLR intracelulares son un evento clave en la patogénesis del LES (fig. 3). Además estos inmunocomplejos pueden activar también monocitos y neutrófilos y así contribuir al proceso inflamatorio y/o vías reguladoras que operan en las enfermedades autoinmunes sistémicas3.
- d)
Producción de INF-α inducida por inmunocomplejos:
Existen diferentes modelos que intentan explicar la formación de inmunocomplejos en un contexto infeccioso. Muchos de los autoantígenos que se generan en este contexto, pueden llegar a ser accesibles al sistema inmune debido a un daño celular excesivo o a una falta de habilidad para eliminar restos apoptóticos77. Por otra parte, los autoanticuerpos que forman parte de estos inmunocomplejos, se pueden producir por células B autorreactivas, que constituyen aproximadamente un 5–20% del repertorio B «naïve» de la mayoría de los individuos78. Además, se sabe que las infecciones virales o bacterianas juegan un papel muy importante en la pérdida de tolerancia y en la producción de autoanticuerpos. De acuerdo con uno de los modelos propuestos70, las DC y los macrófagos (activados a través de sus PRR) pueden regular la expresión de moléculas coestimulatorias y comenzar la producción de INF tipo I y citocinas proinflamatorias. Los autoantígenos que son presentados por estas células pueden estimular potencialmente a células T autorreactivas que promueven la activación, expansión clonal y diferenciación de las célula B autorreactivas, las cuales bajo condiciones apropiadas, pueden sufrir fenómenos de hipermutación somática en su receptor (BcR) y generar inmunoglobulinas más patogénicas. Los clones B autorreactivos pueden ser seleccionados por una mayor cantidad de autoantígenos que se generan en el curso de una infección donde la proporción de muerte celular debida a la acción de células inflamatorias y citotóxicas es mayor. La diferenciación de los linfocitos B también puede estar dirigida por el reconocimiento por parte de linfocitos T de células B infectadas por virus. En cualquiera de los casos, las cantidades excesivas de deshechos celulares y autoanticuerpos que forman inmunocomplejos pueden inducir activación de pDC, vía TLR y establecer un feed-back positivo que exacerba la enfermedad como se muestra en la figura 4.
Un segundo modelo relaciona el proceso de infección a la producción de anticuerpos que reaccionan con autoantígenos por mimetismo molecular. Uno de los principales ejemplos de este fenómeno de crossreactividad, se produce entre péptidos derivados del virus de Epstein-Barr y los autoantígenos Ro (SSA) y Sm, que son proteínas que forman parte de complejos macromoleculares con RNA. Estos complejos son digeridos por células B activadas o células presentadoras de antígeno (APC) que pueden activar un amplio rango de células T autorrectivas y linfocitos B, produciendo un repertorio de autoanticuerpos más extenso.
Una tercera posibilidad es que los linfocitos B autorrectivos se activen a través de sus PRR directamente con el proceso infeccioso mediante señales liberadas por los mismos las cuales unidas a señales del BCR unido a un autoantígeno, que por sí solas estarían por debajo del umbral necesario, serían suficientes para producir este efecto sinérgico entre BCR y TLR, en este contexto79.
- e)
Ligandos internos de los TLR juegan un papel importante en la producción de autoanticuerpos
A pesar de los numerosos mecanismos descritos que pueden provocar la producción de autoanticuerpos cuando hay una infección producida por un microorganismo, también es posible que autoantígenos independientes de procesos de infección, actúen como ligandos endógenos de PRR y tengan un papel activo en la rotura de la tolerancia. Como se ha comentado anteriormente, los TLR7 y TLR9 se localizan en compartimentos citoplasmáticos, por tanto la ruta de activación se limita a moléculas que pueden acceder a esos compartimentos. Una de estas rutas es la endocitosis mediada por el BCR, no solo para microorganismos sino para ligandos endógenos que son reconocidos por linfocitos B autorreactivos, que están normalmente quiescentes.
.")
 inducida por virus, conduce a la producción de autoanticuerpos.")
De forma similar, la expresión aberrante de TLR puede hacer que estas células se hagan hiperrespondedoras a ligandos endógenos y por tanto predispongan a un individuo a desarrollar una enfermedad autoinmune3. Múltiples evidencias en modelos animales soportan esta teoría80,81. Estudios en ratón se han llevado a cabo para evaluar el papel de las respuestas inmunes innatas mediadas por TLR7 y TLR9. Ratones macho BXSP muestran un incremento en la expresión de TLR7 debido a una mutación aceleradora autoinmune ligada al cromosoma Y (Yaa) y desarrollan una enfermedad «lupus-like» con una incidencia mucho más alta que las correspondientes hembras que tienen una expresión normal de TLR782. Por otra parte, la administración de oligodeoxinucleótidos con secuencias inhibitorias puede bloquear la producción de INF-α y en modelos experimentales de LES en ratón, inhibir el desarrollo y la severidad de la enfermedad83.
Existen múltiples evidencias de que numerosos ligandos endógenos pueden estimular otros TLR como TLR2 y/o TLR4. Productos provenientes de la matriz extracelular como el hialuronidato y el heparán sulfato, moléculas liberadas por daño o muerte celular como la HMGB1, fibronectina y proteínas de choque térmico han podido contribuir de manera sustancial en el debut de diferentes enfermedades, aunque este papel, hoy por hoy, no está muy claro.
Lupus Sistémico Eritematoso (LES)Es una enfermedad autoinmune multisistémica crónica que se caracteriza por la producción de autoanticuerpos, en particular contra dsDNA y proteínas nucleares procedentes de muerte celular84. En el hombre, la enfermedad cursa con la afectación de diferentes órganos incluyendo frecuentemente el riñón y el cerebro.
Los pacientes con LES muestran un incremento en la producción de INF-α, que generalmente proviene de las pDC en respuesta a una estimulación continua por inmunocomplejos circulantes, formados principalmente por DNA y RNA85. Como se ha detallado anteriormente, se ha constatado que la internalización de estos inmunocomplejos vía receptores Fc y la subsiguiente detección del DNA y RNA por los TLR 9 y 7 en los compartimentos endosomales de las pDC son un hecho crucial en la patogénesis del LES80,86 (fig. 3)
Diabetes tipo I (T1D)Se trata de una enfermedad autoinmune órgano-específica causada por la destrucción mediada por linfocitos T de las células beta pancreáticas, productoras de insulina, en individuos genéticamente predispuestos84. Actualmente, diversos trabajos han demostrado que las respuestas inmunes innatas mediadas por TLR también podrían contribuir a la inducción de diabetes en ratones. Por ejemplo, células beta apoptóticas son capaces de activar células presentadoras de antígeno vía TLR2 y a su vez estas células pueden activar linfocitos T CD4+ que median la T1D en ratones no obesos (NOD), modelo que desarrolla diabetes espontáneamente con una alta incidencia87.
Esclerosis múltiple (EM)Es una enfermedad desmielinizante del sistema nervioso central que representa una de las mayores causas de parálisis en adultos jóvenes84. El modelo animal de EM, encefalitis autoinmune experimental (EAE), es mediado por linfocitos T CD4+ específicos de mielina, tras inmunización de los animales con proteínas de mielina en presencia de Mycobacterium tuberculosis con adjuvante88. Un cierto número de estudios han documentado que la activación de la inmunidad innata por varios ligandos de TLR es crítica para la activación de las células T CD4+ encefalitogénicas89,90. Linfocitos T de estos ratones pueden transferir la enfermedad a ratones receptores. Se ha observado que la transferencia de estas células T a ratones deficientes de MyD88 provoca una enfermedad muy leve al igual que en receptores deficientes de TLR9, en relación a ratones controles80. Estos datos pueden ser explicados, en parte, por estudios in vitro que muestran que la microglia expresa TLR9 y responde a motivos CpG con la producción de mediadores proinflamatorios77, presumiblemente el ligando en el sistema nervioso central proviene de daño celular producido por células T efectoras patogénicas.
ConclusionesUna de las funciones fundamentales de la respuesta inmune innata es proteger al organismo de patógenos extracelulares e intracelulares e inducir una respuesta inmune adaptativa antígeno-específica contra el organismo invasor. El tipo de respuesta adaptativa que es inducida en el huésped depende fundamentalmente del tipo de patógeno o de varios factores tisulares locales que activan subpoblaciones de células dendríticas vía PRR. Una respuesta inmune innata durante un largo período de tiempo puede romper la tolerancia periférica hacia algunos autoantígenos y resultar en la inducción de enfermedad autoinmune91. Un factor crítico que influye en la susceptibilidad del huésped a desarrollar una enfermedad autoinmune es la respuesta intrínseca de su inmunidad innata a la activación mediada por los TLR. Dependiendo de esta respuesta, las células T autorreactivas pueden diferenciarse en células efectoras y promover el desarrollo de una enfermedad autoinmune. Será crucial, en un futuro el poder dilucidar los mecanismos por los cuales las respuestas inmunes innatas determinan el tipo de inmunidad adaptativa porque ello ayudará a un mejor conocimiento de la patogénesis de las enfermedades autoinmunes. Además, este conocimiento permitirá el desarrollo de nuevas estrategias terapéuticas para combatir este tipo de desórdenes en determinados pacientes92.
Conflicto de interesesLa autora declara no tener ningún conflicto de intereses.
La autora agradece el asesoramiento y las sugerencias recibidas sobre la descripción de las células dendríticas, a los doctores Mar Naranjo Gómez y Francesc Borràs Serres.