




INTRODUCCIÓN
Las enfermedades por depósito lisosómico incluyen un gran número de trastornos caracterizados por defectos en la función lisosómica a raíz de un error congénito del metabolismo1. Los lisosomas son pequeños orgánulos rodeados por una membrana simple que se encuentran dispersos por el citoplasma; contienen hidrolasas ácidas, cuya función es degradar las moléculas complejas de la célula en moléculas más simples. La ausencia de alguna de estas enzimas da lugar a la acumulación progresiva de sustratos que ocasionan una enfermedad por depósito que con frecuencia recibe el nombre del investigador que la describió. Las características clínicas de estas enfermedades dependen de la velocidad y de la intensidad de la acumulación del material que no ha sido degradado y del tipo de sustrato. Son enfermedades de carácter progresivo y multiorgánico, con una gran variabilidad clínica. La identificación del defecto enzimático causante de la enfermedad ha permitido en los últimos años administrar a los pacientes tratamiento enzimático sustitutivo que cuando se realiza en fases tempranas permite cambiar radicalmente el pronóstico de la enfermedad. Sin embargo, al tratarse de enfermedades infrecuentes con expresión clínica muy variable, su diagnóstico puede realizarse tardíamente, cuando el daño es ya irreversible. Muchas enfermedades por depósito lisosómico tienen manifestaciones musculoesqueléticas, a veces en fase temprana, por lo que pueden acudir a una consulta de reumatología. El conocimiento de las características generales de estas enfermedades y de sus manifestaciones reumatológicas resulta fundamental para poder realizar un diagnóstico precoz.
En este trabajo se resumen las características fundamentales de las enfermedades por depósito lisosómico más frecuentes y con manifestaciones musculoesqueléticas, de forma que el reumatólogo pueda familiarizarse con las manifestaciones clínicas y realizar un diagnóstico y tratamiento tempranos.
ENFERMEDADES POR DEPÓSITO LISOSÓMICO. CLASIFICACIÓN Y CARACTERÍSTICAS GENERALES
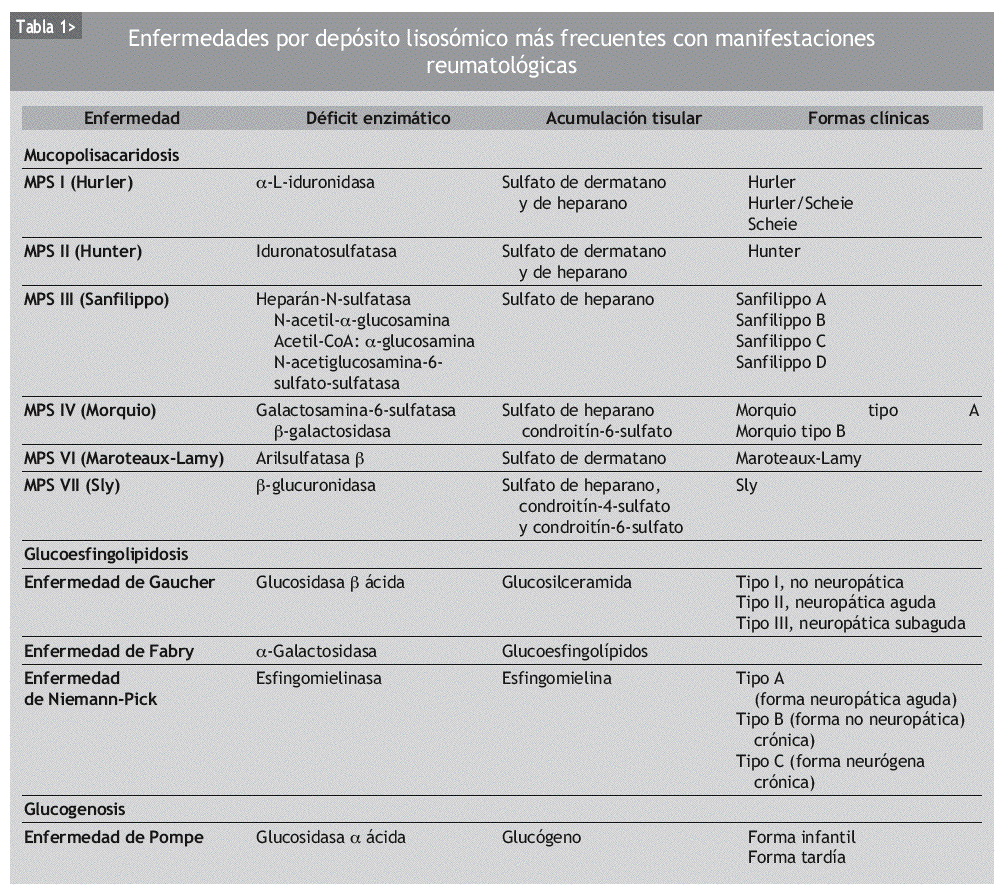
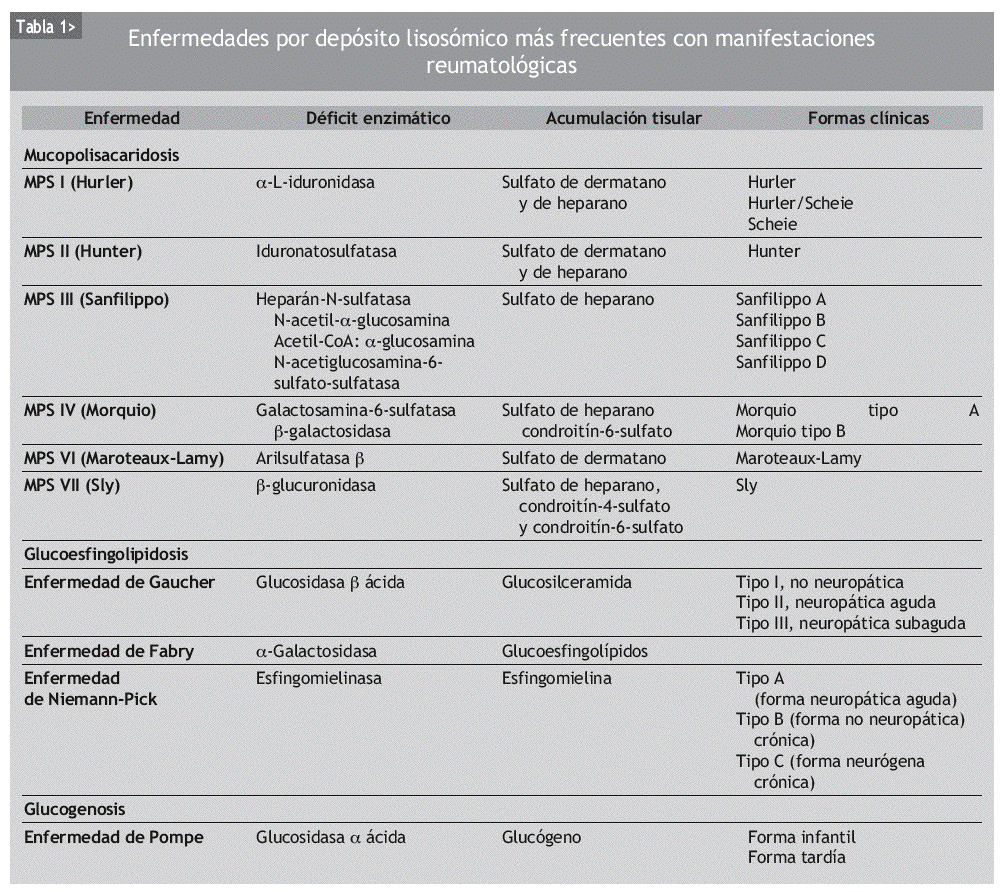
Las enfermedades por depósito lisosómico se clasifican dependiendo de la sustancia acumulada. Aunque el número de enfermedades por depósito lisosómico es elevado, nos centraremos en las que son más frecuentes y/o tienen manifestaciones reumatológicas. En la tabla 1 se presentan las enfermedades por acumulación lisosómica más frecuentes y/o con manifestaciones reumatológicas (mucopolisacaridosis [MPS], enfermedad de Gaucher, enfermedad de Fabry, enfermedad Niemann-Pick y enfermedad de Pompe), si bien existen otras muchas, más infrecuentes y con menos interés desde el punto de vista reumatológico: glucoproteinosis (fucosidosis, manosidosis), mucolipidosis I y II y gangliosidosis.
Las enfermedades por depósito lisosómico son enfermedades infrecuentes, si bien en su conjunto tienen una prevalencia estimada en 1/7.700. De forma individual, la prevalencia es muy inferior (1/100.000 para la MPS I: 2/100.000 para la enfermedad de Gaucher o para la enfermedad de Fabry)2.
Las manifestaciones clínicas de las enfermedades por acumulación lisosómica son muy variables, generalmente de carácter progresivo y multiorgánico: disfunción neurológica progresiva, hepatoesplenomegalia, disostosis esquelética y otras manifestaciones cardiovasculares, respiratorias, dermatológicas y oftalmológicas (tabla 2)3. La herencia es autosómica recesiva, excepto en la enfermedad de Fabry y en la mucopolisacaridosis II (infantil), que están ligadas al cromosoma X. Existe un gran número de mutaciones genéticas. En algunos casos existe relación entre la mutación y la expresión clínica. Existe una alta prevalencia de judíos askenazis (Gaucher, Niemann-Pick, Mucolipidosis, Tay-Sachs). Siempre es necesario realizar un estudio genético al paciente y a los familiares.

El diagnóstico se sospecha ante la presencia de alteraciones óseas, hepatoesplenomegalia, enfermedad neurológica progresiva y otras manifestaciones oftalmológicas, dermatológicas o de otros sistemas. La presencia de antecedentes familiares es un dato importante. Con frecuencia la exploración física facilita una orientación definitiva, como en el caso del aspecto típico de los pacientes con MPS. Para el diagnóstico definitivo es necesario determinar el déficit de la enzima causante de la enfermedad.
La mayor parte de las enfermedades por depósito lisosómico comienzan tempranamente y evolucionan progresivamente con afectación multiorgánica. En los últimos años se ha desarrollado la terapia enzimática de sustitución, que consiste en reemplazar por vía intravenosa la enzima de la que carece el paciente. Existe este tipo de terapia para la MPS I ([laronidasa [Aldurazyme®]), para la MPS-VI ([galsulfasa [Naglazyme®]), la enfermedad de Gaucher (imiglucerasa [Cerezyme®]), la enfermedad de Fabry [agalsidasa b [Fabrazyme®] y agalsidasa a [Replagal®]) y la enfermedad de Pompe (alglucosidasa a [Myozyme®]).
MUCOPOLISACARIDOSIS
Se trata de un grupo de enfermedades debidas a la alteración enzimática en el catabolismo de los glucosaminoglucanos4. Los pacientes con MPS presentan habitualmente una facies tosca típica, visceromegalia, enfermedad ósea (disostosis múltiple), contracturas articulares, retraso metal, cardiopatía, infecciones respiratorias, opacidad corneal, hipoacusia y hernia umbilical e inguinal (tabla 2). La disostosis múltiple se presenta en la MPS I, II, VI y VII. En la MPS III las alteraciones son más leves. En la MPS IV (Morquio) existen alteraciones óseas con características diferenciales. La gravedad depende de la cantidad se glucosaminoglucanos acumulados en relación con el grado de deficiencia enzimática, que a su vez está en relación con el genotipo del paciente. La prevalencia es de 1/20.000 nacidos vivos. Aunque la mayor parte de las características clínicas de la MPS son comunes, cada una tiene aspectos clínicos diferenciales que comentaremos brevemente. La determinación de los glucosaminoglucanos en orina y la determinación de la actividad de la enzima causante de la enfermedad permite el diagnóstico definitivo.
MPS I
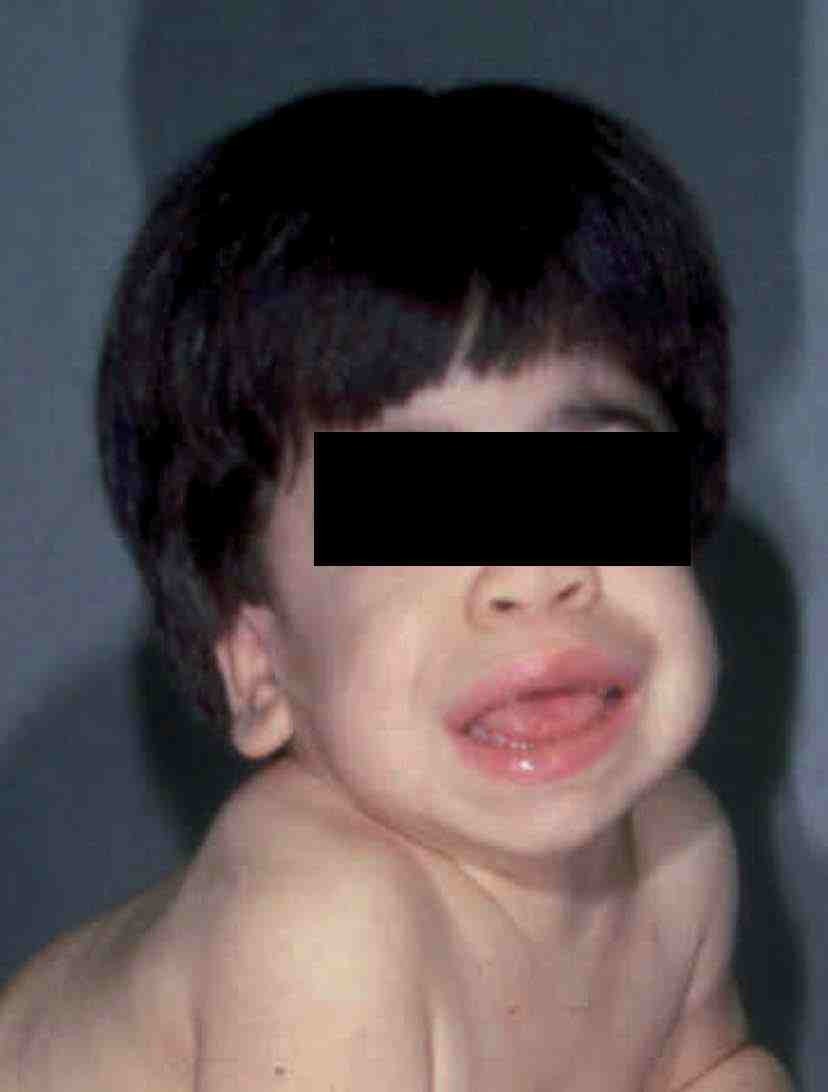
La causa de la MPS I es un déficit de *-L-iduronidasa que da lugar a una acumulación de sulfato de dermatano y de heparano. Se debe a una mutación del gen localizado en el cromosoma 4p16.3. Existen 3 tipos de MPS I: la enfermedad de Hurler (forma grave), la enfermedad de Hurler/Scheie (forma moderada) y la enfermedad de Scheie (forma atenuada) (tabla 2). Los pacientes con enfermedad de Hurler comienzan en la infancia, entre los 6 meses y 2 años de edad, con infecciones respiratorias de repetición, retraso mental, visceromegalia, cardiopatía, contracturas articulares, enfermedad ósea, hernias umbilicales e inguinales, opacidad corneal y la típica facies5 (fig. 1). Generalmente mueren antes de los 10 años. Los pacientes pueden presentar también alteraciones craneofaciales con agrandamiento de la silla turca, malformaciones dentarias e hidrocefalia.
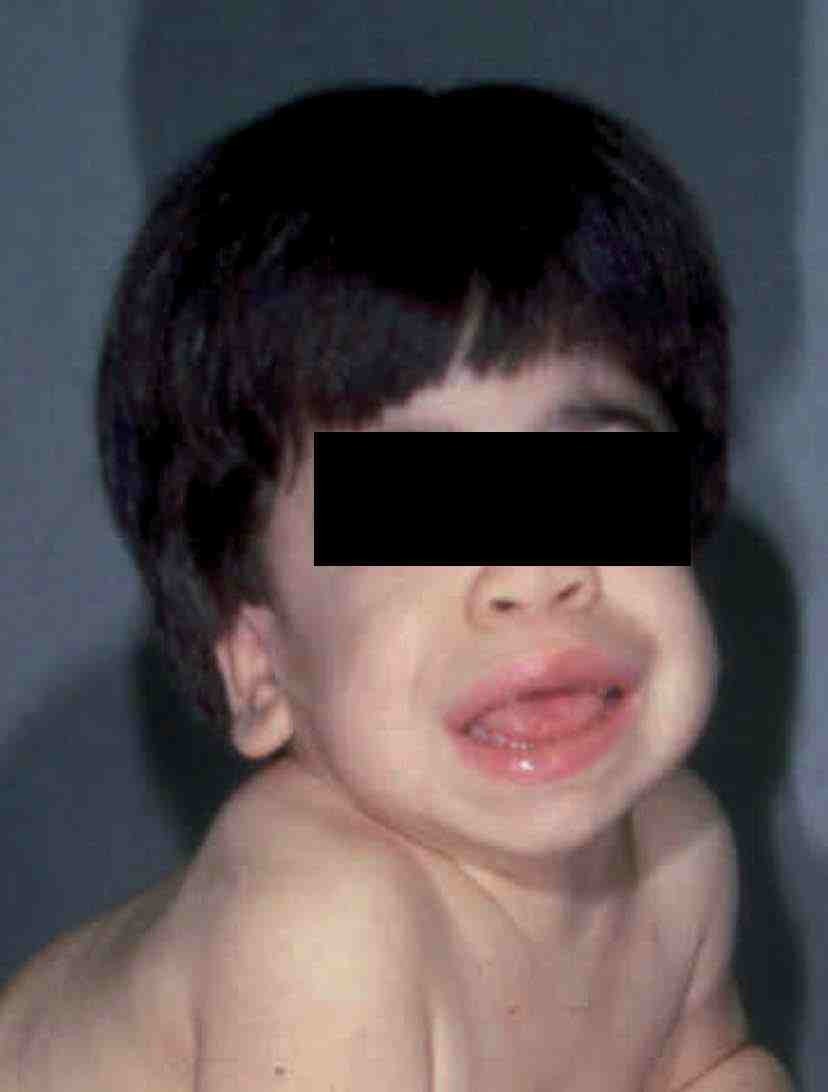
Figura 1 Paciente con MPS I (enfermedad de Hurler). (Cortesía del Dr. E. Kakkis.)
La enfermedad de Hurler/Scheie se presenta entre los 2 y los 6 años de edad y tiene una gravedad moderada, a veces sin retraso mental. El paciente con enfermedad de Hurler/Scheie presenta los aspectos faciales típicos de las MPS, pero menos marcados que los pacientes con enfermedad de Hurler. Generalmente tienen contracturas articulares, enfermedad ósea y visceromegalia. Los pacientes generalmente mueren durante la tercera década por enfermedad cardíaca (valvulopatía) o respiratoria.
La enfermedad de Scheie es la forma atenuada de la MPS I, y aunque comparte las manifestaciones clínicas de la enfermedad de Hurler/Scheie, las manifestaciones comienzan habitualmente en la segunda década y los pacientes pueden vivir hasta la edad adulta. Son pacientes especialmente difíciles de diagnosticar, sin el aspecto típico de la MPS (fig. 2), y en ocasiones presentan únicamente manifestaciones musculoesqueléticas, contracturas articulares y síndrome del túnel carpiano. También pueden desarrollan displasia de cadera, valvulopatía cardíaca y opacidad corneal.

Figura 2 Paciente con MPS I. Forma atenuada (enfermedad de Scheie). (Cortesía de la Dra. N. Guffon.)
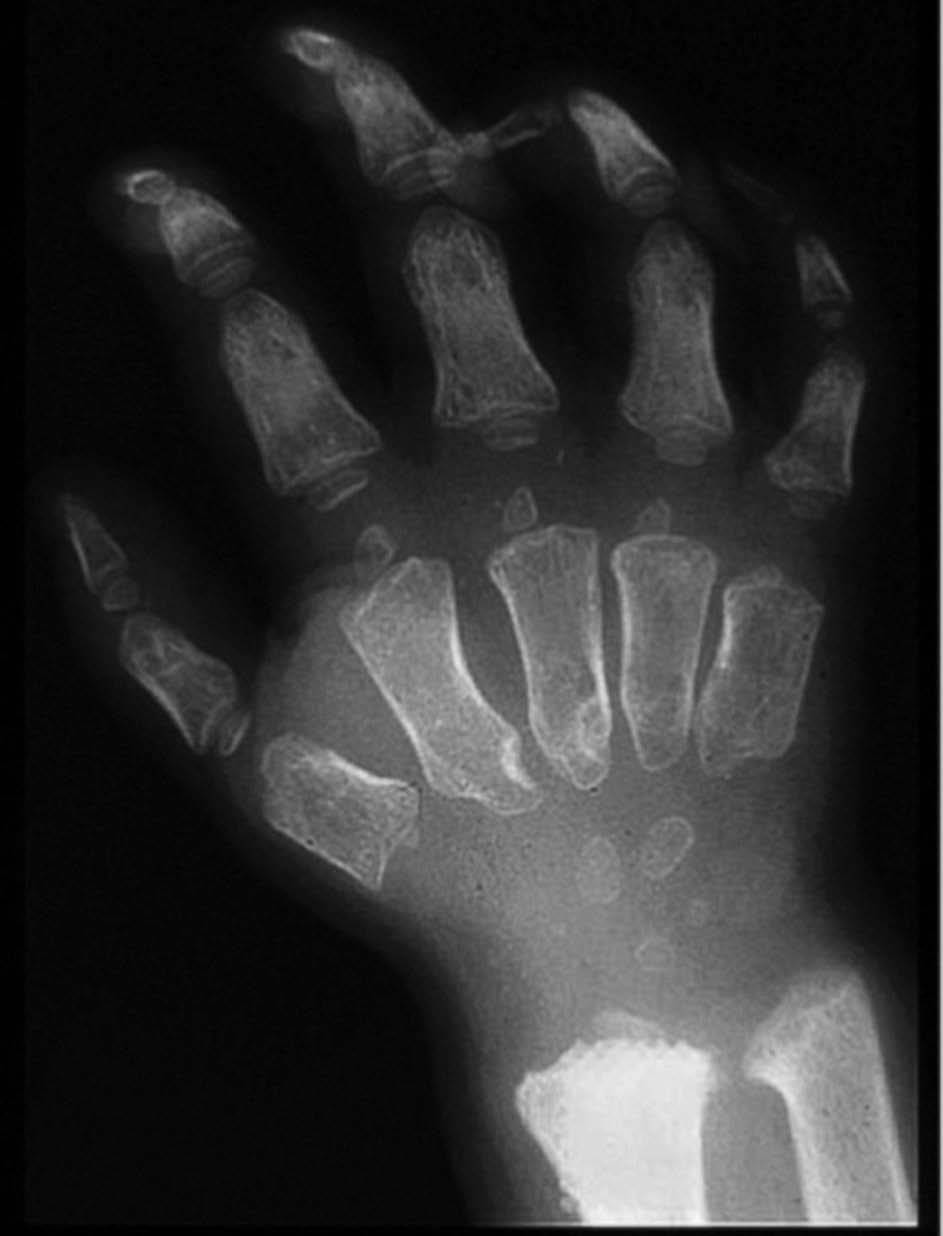
Las manifestaciones reumatológicas de los pacientes con MPS I son la disostosis múltiple, el síndrome del túnel carpiano y las contracturas articulares. La disostosis múltiple es común a todas las MPS y se caracteriza por la presencia de múltiples alteraciones en el esqueleto axial y periférico6,7. En la columna los pacientes presentan alteración en el desarrollo de la porción anterosuperior de los cuerpos vertebrales con cifosis (típica giba). Los cuerpos vertebrales tienen un aspecto oval con disminución de la altura. Se observa también subluxación vertebral y cifoescoliosis. Es frecuente la coxa valga con dislocación de la cadera y displasia de la cabeza femoral. Se observa ensanchamiento de la parte anterior de la cortical y la porción medial de la clavícula. En los huesos tubulares largos se aprecia expansión diafisaria y metafisaria con retardo en la osificación epifisaria. En las manos se aprecia osteopenia, hipoplasia del carpo y deformidad de la porción anterior del cúbito y del radio, las falanges proximales y medias son anchas y están acortadas, y las falanges terminales y los huesos del carpo son hipoplásicos (fig. 3).
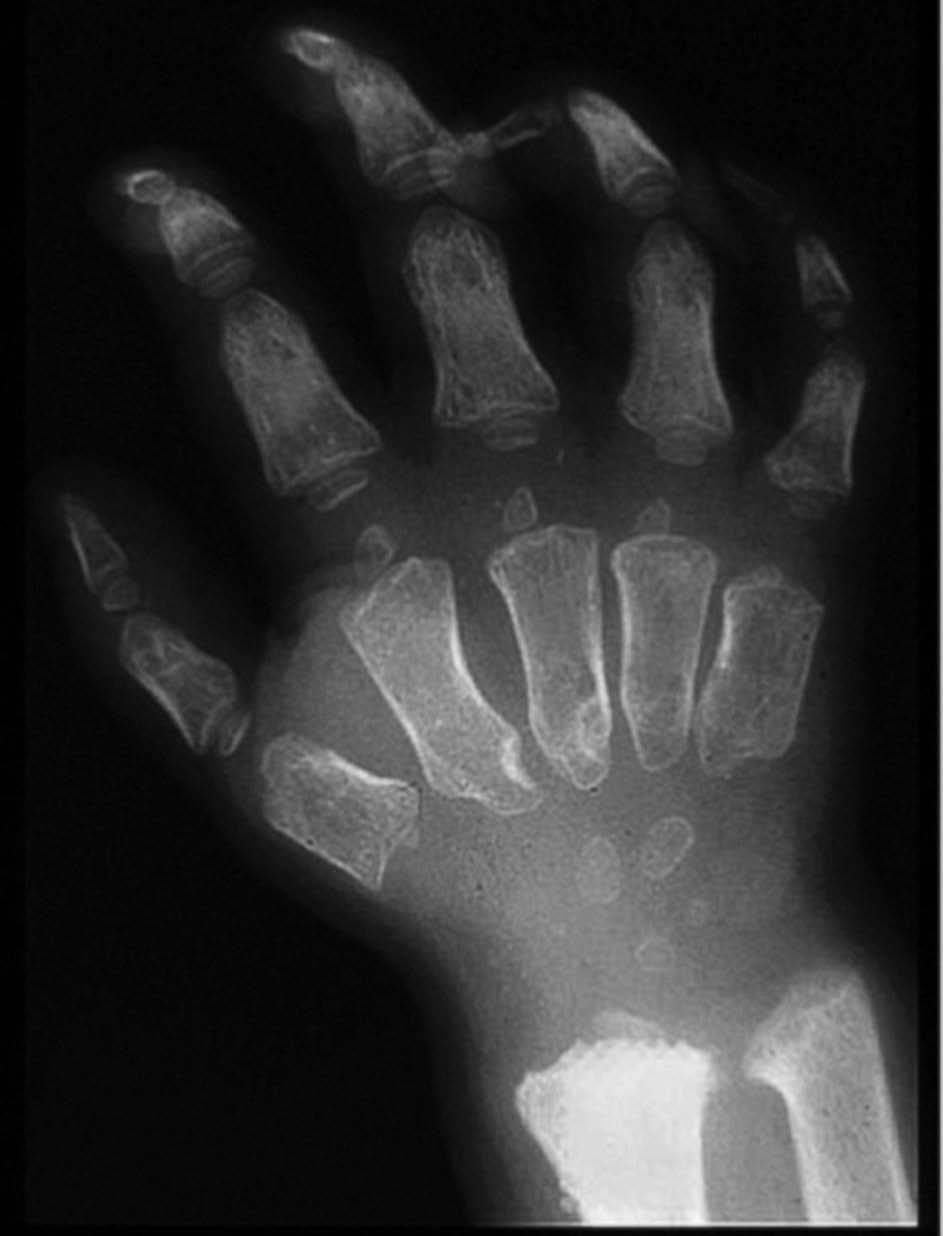
Figura 3 MPS I. Osteopenia, ensanchamiento de los huesos tubulares, hipoplasia del carpo y deformidad en V de la porción distal de cúbito y radio. (Cortesía del Dr. Wraith.)
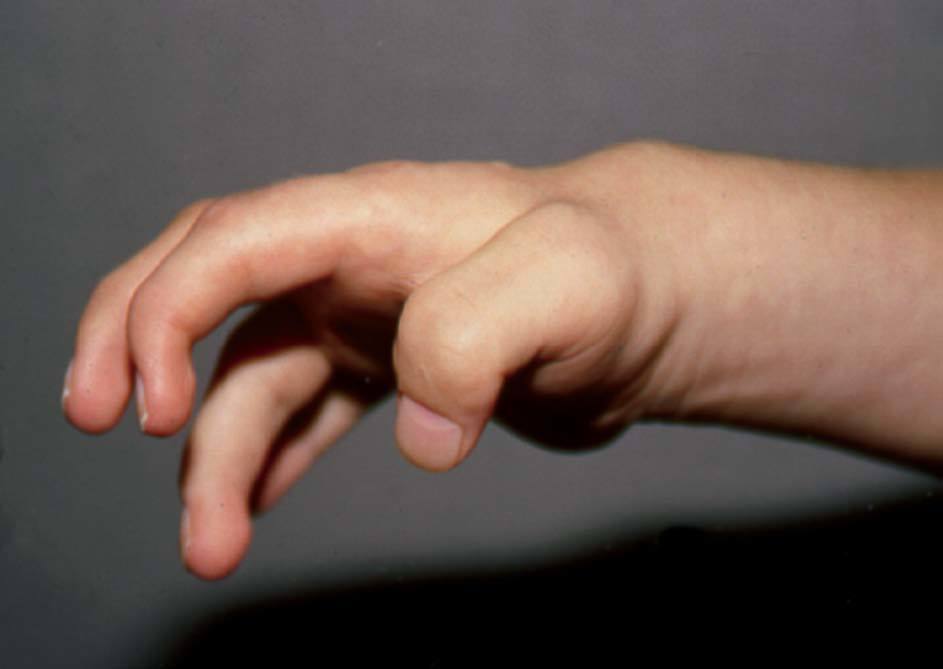
La infiltración de mucopolisacáridos en tendones, en ligamentos y en la cápsula articular ocasiona contracturas articulares simétricas en hombros, codos, manos, caderas y rodillas, con dolor articular y limitación funcional progresiva. Los pacientes desarrollan rigidez articular con contracturas articulares progresivas con dolor8. Las contracturas en las manos, junto con las alteraciones propias de la disostosis múltiple, dan lugar a la típica mano en garra de estos pacientes (fig. 4).
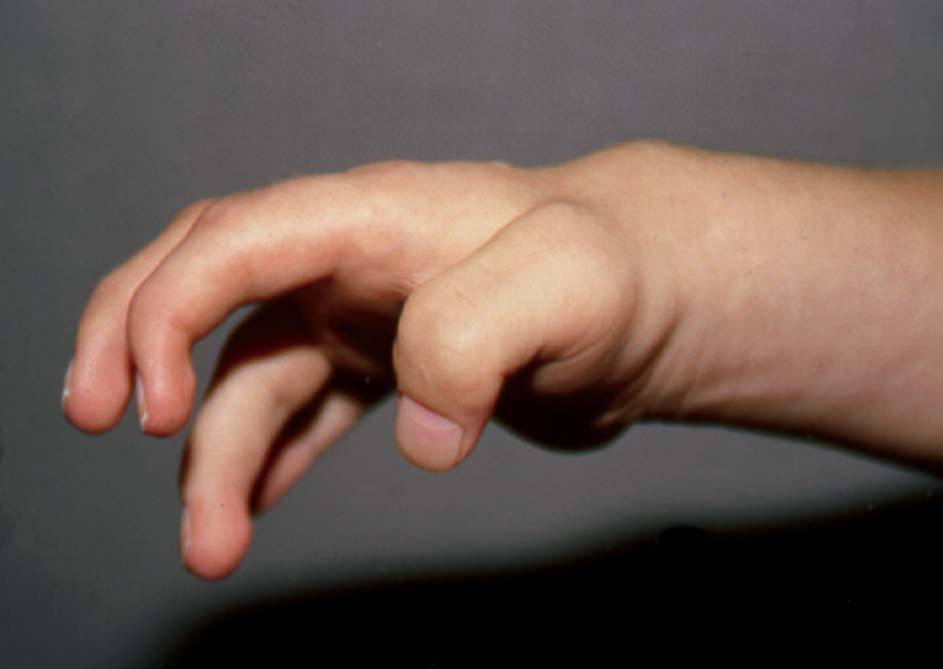
Figura 4 MPS I. Mano en garra. (Cortesía de la US MPS Society.)
Cuando existe un síndrome del túnel carpiano, los pacientes no tienen dolor o alteraciones sensitivas hasta que la lesión del nervio está muy evolucionada. Sin embargo, son muy frecuentes la atrofia de la eminencia tenar y la pérdida de fuerza. El síndrome de túnel carpiano es raro en niños, si bien es muy frecuente en la MPS.
El trasplante de médula ósea se ha usado con éxito en el tratamiento de la MPS I, aunque en el momento actual el tratamiento consiste en la administración de laronidasa (Aldurazyme®), que reduce la hepatoesplenomegalia a la vez que mejora la movilidad articular y la función cardíaca9.
MPS II (HUNTER)
El déficit de iduronatosulfatasa produce una acumulación de sulfato de dermatano y de heparano. Esta alteración enzimática se debe a una alteración del cromosoma Xq28 y se denomina enfermedad de Hunter. Aunque esta enfermedad está ligada al cromosoma X, se han descrito casos en mujeres. Las características clínicas son similares al Hurler: facies tosca, hepatoesplenomegalia, retraso mental, disostosis ósea, cardiopatía y sordera, si bien no presentan opacidad corneal y el comienzo es más tardío, con supervivencia hasta la segunda y la tercera décadas de la vida. Algunos pacientes presentan típicas lesiones nacaradas en la espalda, en los brazos y en los muslos. Existen formas leves con características parecidas a la enfermedad de Scheie. La Food and Drug Administration (FDA) estadounidense aprobó en julio de 2006 el tratamiento enzimático sustitutivo con idursulfasa para la MPS II en dosis de 0,5 mg/kg, administrado semanalmente.
MPS III (SANFILIPPO)
La MPS III (enfermedad de Sanfilippo) de debe al déficit de varias enzimas implicadas en la degradación del sulfato de heparano que permiten clasificar a los pacientes en 4 formas (A, B, C y D) similares desde el punto de vista clínico, si bien la forma A es la más grave. Las manifestaciones clínicas comienzan en la primera década de la vida con retraso mental progresivo y grave, hepatoesplenomegalia, típico aspecto facial y contracturas articulares. Los hallazgos físicos son menos marcados que en la enfermedad de Hurler. Las alteraciones óseas por disostosis múltiple son leves o moderadas. Algunos pacientes viven hasta la edad adulta.
MPS IV (MORQUIO)
La enfermedad de Morquio (MPS IV) se debe a la deficiencia de 2 enzimas (galactosamina-6-sulfatasa y ß-galactosidasa), que ocasiona una acumulación de sulfato de heparano (Morquio tipo A) y 6 sulfato de condroitina (Morquio tipo B). El aspecto más característico de la enfermedad de Morquio es el desarrollo de enfermedad ósea grave sin retraso mental10. Generalmente presentan facies tosca, hepatoesplenomegalia, enfermedad valvular cardíaca y opacidad corneal. Los pacientes desarrollan acortamiento de la estatura, disostosis múltiple con displasia espondiloepifisaria y platispondilia, marcada hiperlordosis lumbar y laxitud articular, así como coxa valga con displasia de la cabeza femoral. La laxitud articular es un aspecto típico de la enfermedad de Morquio, a diferencia de las contracturas articulares que se presentan en otras MPS. Es típica la deformidad en genu valgum y contractura en flexión. La hipoplasia de la apófisis odontoides puede ocasionar subluxación C1-C2 y es una característica típica de la enfermedad de Morquio. Algunos pacientes con formas leves pueden alcanzar la edad adulta.
MPS VI (MAROTEAUX-LAMY)
El déficit de arilsulfatasa ß da lugar a la MPS VI (enfermedad de Maroteaux-Lamy), en la que se acumula sulfato de dermatano. Los pacientes no tienen retraso mental, pero presentan facies tosca, hepatoesplenomegalia, opacidad corneal y disostosis múltiple. Los pacientes adolescentes o adultos pueden presentar una miocardiopatía grave o una enfermedad valvular. Pueden desarrollar estenosis del canal cervical. En las formas graves los pacientes mueren en la segunda o la tercera década de la vida. Existen formas leves similares a la enfermedad de Scheie. La galsulfasa (Naglazyme®) es el tratamiento enzimático sustitutivo comercializado para la MPS VI.
MPS VII (ENFERMEDAD DE SLY)
Los pacientes con MPS VII se caracterizan por la acumulación de sulfato de heparano, condroitín-4-sulfato y condroitín-6-sulfato. Las manifestaciones clínicas son similares a las de la MPS I (Hurler), con importantes alteraciones óseas, facies típica y visceromegalia. Las formas moderadas comienzan tardíamente y pueden no tener retraso mental.
GLUCOESFINGOLIPIDOSIS
Enfermedad de Gaucher
La enfermedad de Gaucher se debe al déficit de glucosidasa ß ácida, que ocasiona una acumulación de glucosilceramida, fundamentalmente en el hueso, el hígado, el bazo y el sistema nervioso central. El gen que codifica esta enzima se localiza en el cromosoma 1q21, y se conocen cerca de 200 mutaciones del gen. En los pacientes con enfermedad de Gaucher se acumula glucosilceramida en los macrófagos (células de Gaucher). Los pacientes presentan hepatoesplenomegalia, anemia, trombopenia, fatigabilidad intensa y manifestaciones óseas (tabla 2). La enfermedad se presenta en 1/40.000 nacidos vivos11. Existen 3 formas, y la más frecuente es la tipo I (no neuropática). Los pacientes con las formas II y III son menos y se caracterizan por la afectación neurológica. El tipo II (neuropática aguda) comienza antes de los 2 años, con enfermedad neurológica grave y fallecimiento temprano. El tipo III (neuropática subaguda) se inicia en la infancia o en la adolescencia y cursa con demencia y ataxia. La enfermedad tiene una prevalencia alta entre judíos askenazis.
La enfermedad Gaucher tipo I constituye el 90% de los pacientes. El comienzo de las manifestaciones clínicas es muy variable, si bien la mayor parte de los casos se diagnostican entre al segunda y la tercera década. Los pacientes desarrollan hepatomegalia y esplenomegalia masiva. La anemia y la trombocitopenia están en relación con la infiltración de la médula ósea y el secuestro esplénico de plaquetas. Los pacientes presentan episodios de sangrado y fatigabilidad intensa. Puede haber afectación pulmonar intersticial e hipertensión pulmonar. La enfermedad de Gaucher es habitualmente progresiva, especialmente si comienza en la infancia12.
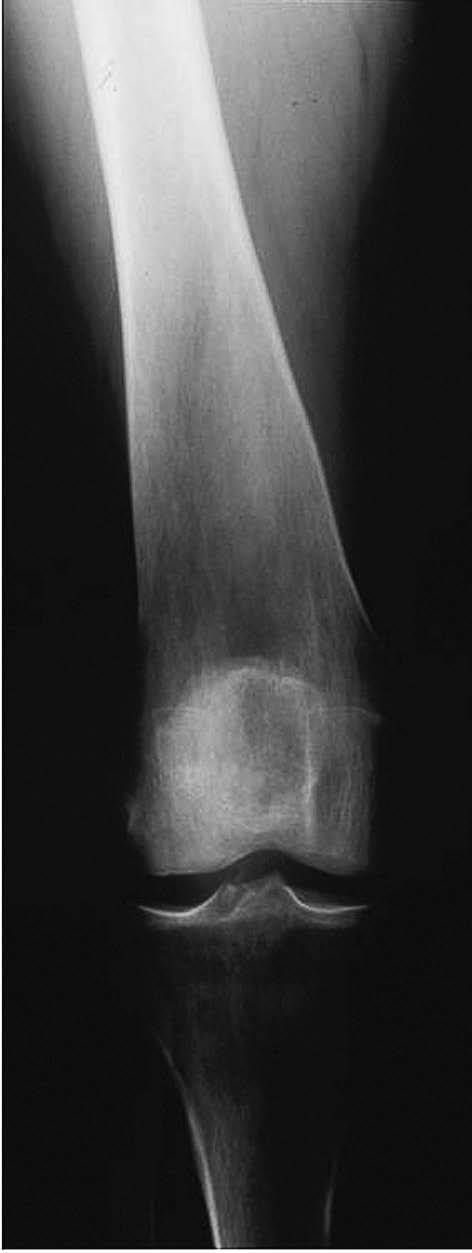
La afectación ósea puede ser muy importante y grave, con diversas manifestaciones. Se debe a la infiltración de macrófagos cargados de lípidos (células de Gaucher) y ocasiona osteopenia con remodelamiento del contorno óseo con la típica deformidad en matraz de Erlenmeyer (fig. 5). Puede haber fracturas espontáneas u osteonecrosis13. Generalmente existe retraso del crecimiento. Los pacientes tienen dolor óseo crónico o agudo por alguna de las complicaciones descritas, así como episodios de dolor óseo intenso con fiebre. Pueden presentar artralgias intensas con tumefacción articular y derrame14.
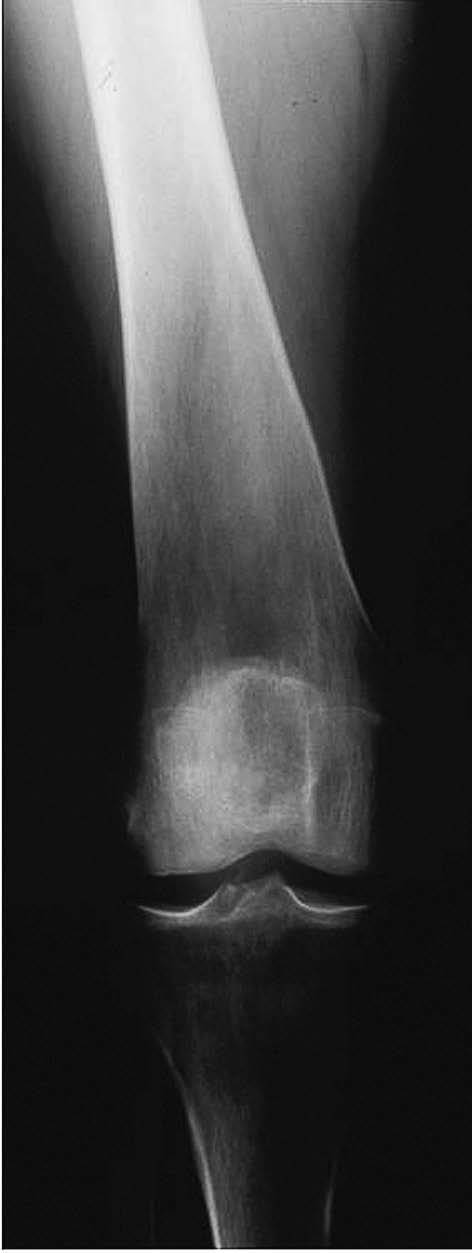
Figura 5 Enfermedad de Gaucher. Deformidad de Erlenmeyer. (Cortesía del Dr. L. W. Poll.)
El curso clínico es muy variable, desde una enfermedad muy grave en niños hasta un curso crónico y moderado en adultos. Los pacientes pueden morir por sangrado, enfermedad pulmonar o infecciones. Existe una mayor incidencia de cáncer15.
El tratamiento enzimático de sustitutivo se debe instaurar en pacientes sintomáticos con malnutrición, alteración del crecimiento y limitación funcional con recuento plaquetario por debajo de 60.000/ml, hepatomegalia superior a 2,5 veces el tamaño normal, esplenomegalia superior a 15 veces el tamaño normal y evidencia radiológica de enfermedad ósea16. El tratamiento con imiglucerasa, 30-60 U/kg por vía intravenosa cada 2 semanas, permite disminuir la anemia y la trombocitopenia, disminuir el crecimiento del hígado y del bazo, eliminar el dolor óseo, prevenir el desarrollo de fracturas y osteonecrosis, aumentar la masa ósea, normalizar el crecimiento y mejorar la calidad de vida17. La dosis se debe individualizar, dependiendo de las características clínicas de cada paciente. Los objetivos de tratamiento son incrementar la hemoglobina a 11 g/dl a los 12-24 meses de tratamiento, y para la trombocitopenia aumentar la cifra de plaquetas 1,5-2 veces durante el primer año y normalizar la cifra durante el segundo (para las trombocitopenias moderadas) o entre el segundo y el quinto año para las graves. La hepatomegalia y la esplenomegalia se deben reducir, manteniendo el volumen como máximo entre 1 y 5 veces el valor normal. Se debe eliminar el dolor óseo entre 1 y 2 años, evitar las crisis de dolor óseo, evitar la osteonecrosis y alcanzar valores normales de masa ósea. En los niños se debe alcanzar la talla normal para su edad en 3 años y alcanzar el desarrollo puberal normal11.
Enfermedad de Fabry
La enfermedad de Fabry se debe a la deficiencia de *-galactosidasa, que ocasiona una acumulación de glucoesfingolípidos en el endotelio, el riñón, el corazón y el sistema nervioso periférico. Se transmite de forma recesiva ligado al cromosoma X y se presenta en 1/40.000 varones.
Los pacientes comienzan generalmente en la segunda década de la vida con una neuropatía periférica con acroparestesias y episodios agudos de dolor. Desarrollan angioqueratomas (fig. 6), opacidades corneales (córnea verticillata), hipohidrosis y daño vascular progresivo, que afecta al riñón (insuficiencia renal), al corazón (hipertrofia ventricular izquierda) y al sistema nervioso (accidentes cerebrovasculares y otras múltiples manifestaciones neurológicas). El paciente con enfermedad de Fabry desarrolla de forma progresiva insuficiencia renal y enfermedad vascular periférica, que puede ocasionar la muerte18-21.
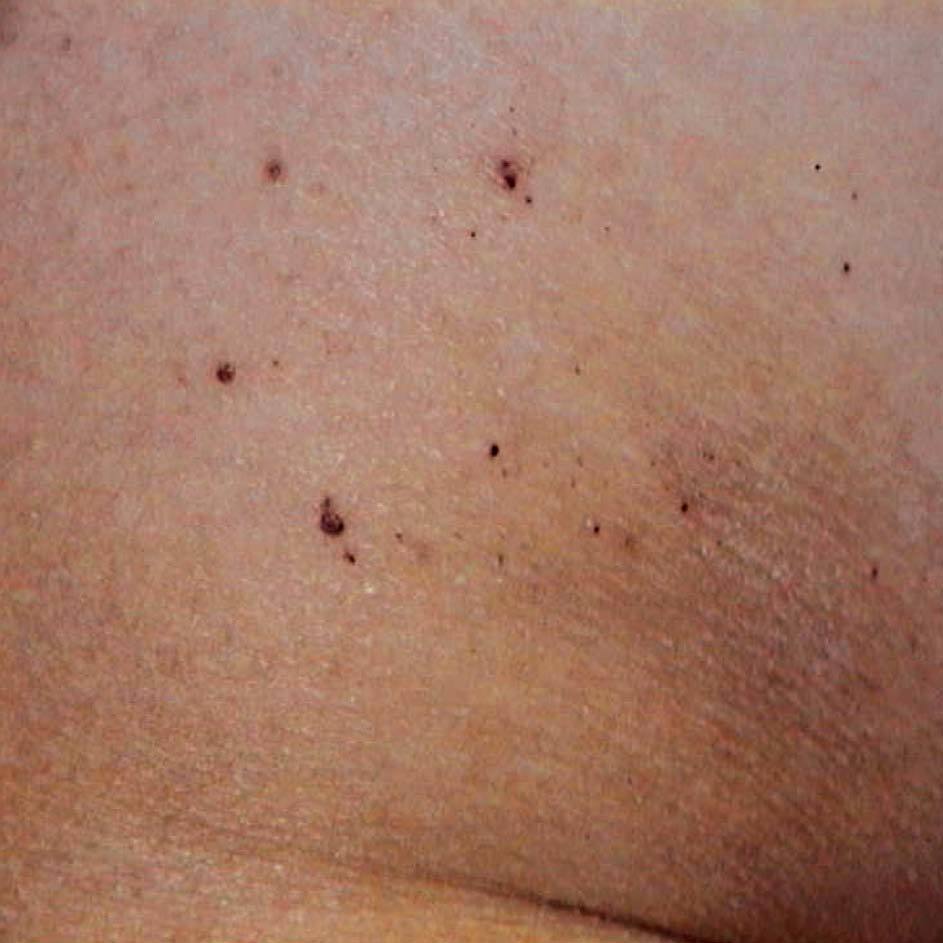
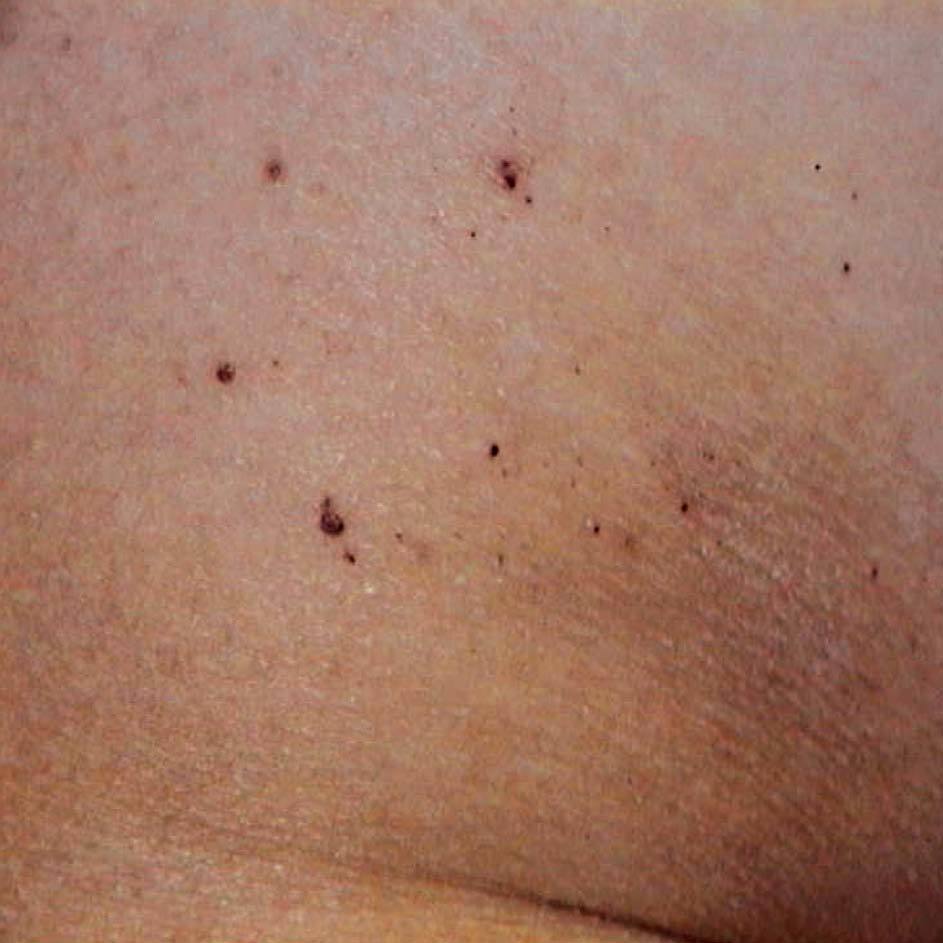
Figura 6Enfermedad de Fabry. Angioqueratomas.
El paciente con enfermedad de Fabry puede llegar a una consulta de reumatología refiriendo acroparestesias con dolor y sensación de quemazón en las manos y los pies, episodios de dolor intenso osteomuscular, desencadenados por estrés, por el calor o al hacer deporte, en ocasiones acompañados de fiebre. También pueden desarrollar manifestaciones de entesopatía, presentar fracturas y osteonecrosis.
El diagnóstico se realiza demostrando disminución de la actividad de la *-galactosidasa en el plasma o en los leucocitos. Se puede realizar el diagnóstico prenatal con demostración de la alteración enzimática en cultivo de células amnióticas. Se puede valorar la posibilidad de cribado en pacientes con hipertrofia ventricular o insuficiencia renal de causa desconocida, teniendo en cuenta que existe un tratamiento eficaz para estos pacientes.
El tratamiento consiste en la administración de tratamiento enzimático sustitutivo con agalsidasa ß (Fabrazyme®), a una dosis de 1 mg/kg por vía intravenosa cada 2 semanas22, o con agalsidasa a (Replagal®), a 0,2 mg/kg cada 2 semanas23.
Enfermedad de Niemann-Pick
Los pacientes con enfermedad de Niemann-Pick tienen un déficit de esfingomielinasa ácida que ocasiona una acumulación de esfingomielina. Es más prevalente en judíos askenazis. La enfermedad de Niemann-Pick tipo A (forma neuropática aguda) se presenta en los primeros meses de la vida con hepatoesplenomegalia y grave disfunción neurológica (ataxia, distonía, convulsiones) que ocasiona la muerte. La enfermedad de Niemann-Pick tipo B (forma crónica no neuropática) comienza en la infancia y tiene un curso crónico hasta la edad adulta con hepatoesplenomegalia, alteraciones oculares, alteración de la función pulmonar, cardiopatía y alteraciones óseas. Existe anemia e hipercolesterolemia con lipoproteínas de baja densidad24. La enfermedad de Niemann-Pick tipo C (forma neurógena crónica) comienza tardíamente en la infancia con una afectación neurológica grave que habitualmente ocasiona la muerte en la segunda década de la vida. El diagnóstico se realiza mediante estudio del cultivo de fibroblastos, que muestran acumulación de lípidos intracelulares.
Las manifestaciones reumatológicas de la enfermedad de Niemann-Pick se deben al infiltrado de la médula ósea por macrófagos cargados de lípidos. Los pacientes presentan dolor óseo y alteraciones radiológicas caracterizadas por osteopenia, ensanchamiento medular, trabeculación grosera y alteraciones en el contorno óseo (deformidad de Erlenmeyer). En la actualidad, un ensayo clínico en fase II valora la eficacia y la seguridad de un tratamiento enzimático sustitutivo para pacientes con Niemann-Pick tipo B.
GLUCOGENOSIS
Enfermedad de Pompe
En los pacientes con enfermedad de Pompe se produce una acumulación de glucógeno secundario a la deficiencia de glucosidasa a ácida (tabla 1). La forma infantil se presenta tempranamente con hipotonía, miocardiopatía con insuficiencia cardíaca y fallecimiento durante la infancia (tabla 2). En la forma juvenil y del adulto los pacientes presentan debilidad muscular progresiva de predominio en cintura escapular y pelviana. Puede ocasionar insuficiencia respiratoria. El electromiograma muestra un patrón miopático, y las enzimas musculares están elevadas. La biopsia muscular muestra una miopatía vacuolar con acumulación citoplasmática de glucógeno25. El diagnóstico se puede confirmar comprobando la deficiencia de glucosidasa a ácida en linfocitos o en fibroblastos obtenidos mediante biopsia de piel.
El tratamiento enzimático sustitutivo se realiza mediante la administración intravenosa de alglucosidasa * (Myozyme®) en dosis 20 mg/kg cada 2 semanas.
OTRAS ENFERMEDADES POR DEPÓSITO LISOSÓMICO
Debemos conocer también qué otras enfermedades por acumulación lisosómica, aunque más infrecuentes, pueden presentar manifestaciones reumatológicas. Es recomendable conocer alguno de sus aspectos, puesto que tienen características comunes (en algunos casos son muy similares) con las mucopolisacaridosis, especialmente con el tipo I (Hurler) y II (Hunter)1.
En el grupo de las glucoproteinosis están la fucosidosis y la manosidosis. La fucosidosis de debe a la deficiencia de la enzima *-fucosidasa. Los tipos I y II comienzan en los primeros años de vida con deterioro neurológico y muerte temprana. El tipo III comienza tardíamente. Los pacientes presentan alteraciones de disostosis múltiple. La manosidosis se debe a una deficiencia de la *-D-manosidasa, y presenta 2 formas clínicas. La forma infantil se asocia con retraso mental y graves alteraciones óseas, y la forma juvenil cursa con cambios moderados de disostosis múltiple.
Las mucolipidosis (la I y la II) son enfermedades que se parecen clínicamente a la enfermedad de Hurler. Se describen 4 formas clínicas (la tipo IV no presenta alteraciones óseas). La mucolipidosis tipo I (sialidosis) se debe a una deficiencia de la N-acetil neuraminidasa y se manifiesta con un fenotipo similar al Hurler, hepatoesplenomegalia, enfermedad cerebral neurodegenerativa y múltiples alteraciones esqueléticas: escoliosis toracolumbar, dislocación de caderas, adelgazamiento de los huesos tubulares e hipoplasia de odontoides. La mucolipidosis tipo II (síndrome de Leroy) en la forma neonatal puede confundirse con hiperparatiroidismo o con osteomalacia, y se observa osteopenia, aposición perióstica, fracturas, cuerpos vertebrales ovoides y giba toracolumbar. Existe una forma de comienzo tardía en la que los pacientes presentan disostosis múltiple. La mucolipidosis tipo III (polidistrofia seudo Hurler) cursa con retraso mental, retardo del crecimiento y disostosis múltiple, con manifestaciones clínicas que simulan una MPS tipo I y II.
La gangliosidosis GM1 se debe a una deficiencia de ß-galactosidasa y cursa con visceromegalia, degeneración neurológica, disostosis múltiple con un aspecto similar al Hurler. Se describen 3 formas. La infantil provoca graves alteraciones neurológicas, ceguera, hepatoesplenomega CLASS=01Ladillo> MANIFESTACIONES REUMATOLÓGICAS DE LAS ENFERMEDADES POR DEPÓSITO LISOSÓMICO Y DIAGNÓSTICO DIFERENCIAL
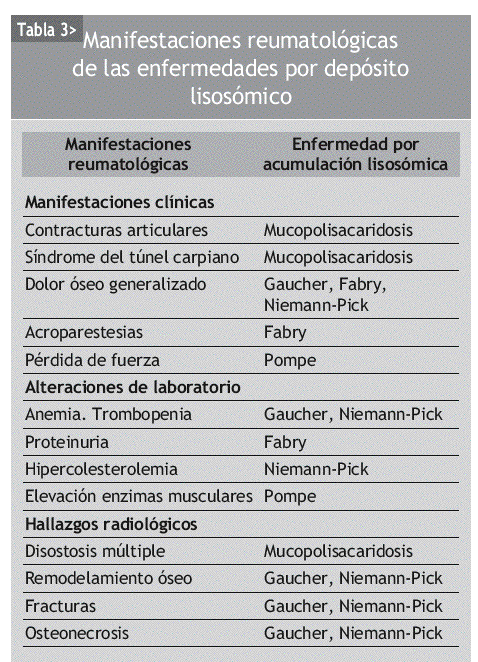
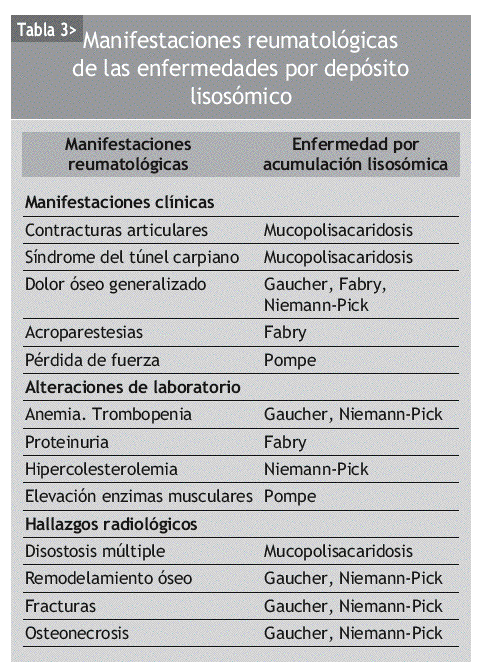
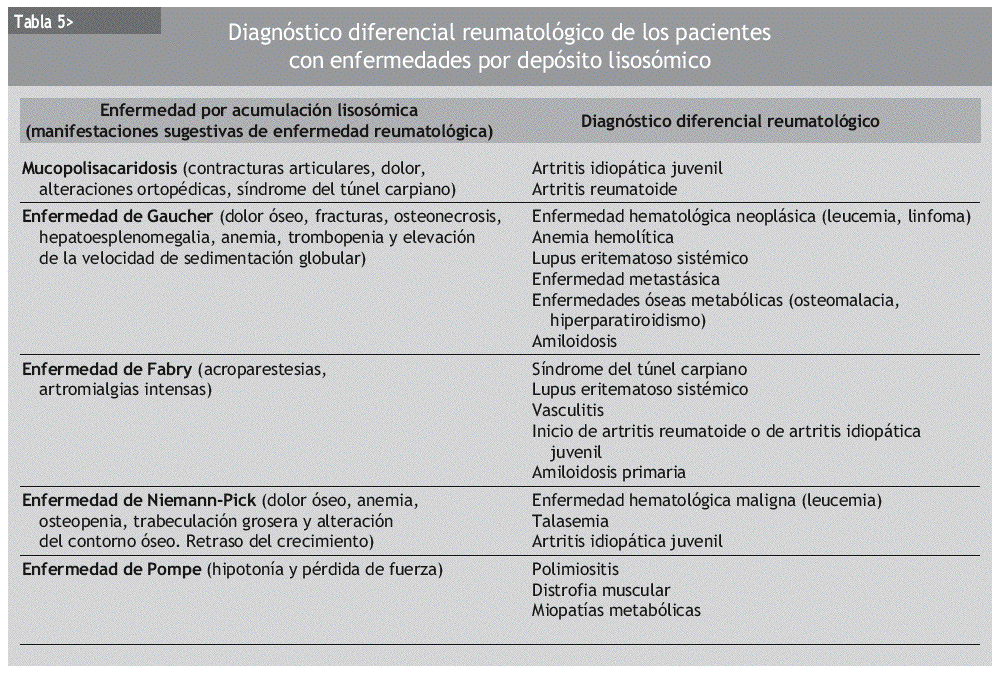
Las manifestaciones reumatológicas son muy frecuentes en las enfermedades por acumulación lisosómica. La presencia de alguna de las manifestaciones que se describen en la tabla 3 debe hacer pensar al reumatólogo en la posibilidad de alguna de estas enfermedades. Sin duda, la presencia de otras manifestaciones sistémicas en el paciente puede ser la clave que oriente al diagnóstico (tabla 2).
En los pacientes con MPS con formas graves, comienzo temprano y expresión clínica completa, la sospecha del diagnóstico es fácil, especialmente por sus características físicas típicas. Sin embargo, las formas leves y de comienzo tardío pueden ser muy difíciles de diagnosticar y hay que consultar a un reumatólogo, si se tiene en cuenta lo frecuentes que son las manifestaciones musculoesqueléticas en estos pacientes. Los pacientes con formas moderadas (Hurler/Scheie) o atenuadas (Scheie) de MPS I, así como con formas leves de la MPS II (Hunter) y de MPS VI (Maroteaux-Lamy), pueden presentar únicamente un síndrome del túnel carpiano, alteraciones ortopédicas como displasia de cadera y contracturas articulares con dolor, con un aspecto físico normal y sin retraso mental. En estos pacientes es posible que el diagnóstico inicial del reumatólogo sea de una enfermedad inflamatoria de mecanismo inmunitario, como una artritis idiopática juvenil o una artritis reumatoide. No hay duda que una mano en garra, como la de la figura 3, puede plantear muchas dudas en el diagnóstico. Sin embargo, la ausencia de artritis, la negatividad de parámetros de laboratorio propios de la artritis idiopática juvenil o de la artritis reumatoide y la presencia de alguna otra manifestación de la MPS (opacidad corneal, cardiopatía) pueden hacer pensar en una MPS.
Los pacientes con enfermedad de Gaucher presentan habitualmente fatigabilidad progresiva junto con dolor óseo y articular, con brotes de dolor intenso que pueden acompañarse de fiebre. Puede detectarse hepatoesplenomegalia, anemia y trombopenia. Es preciso descartar una enfermedad neoplásica, tanto hematológica (leucemia, linfoma) como metastásica. La drepanocitosis puede producir dolor óseo, anemia y osteonecrosis. Un paciente con trombopenia, esplenomegalia y artralgias puede hacer pensar en un lupus. Los pacientes con osteomalacia también tienen dolor óseo, osteopenia y fracturas. Sin embargo, la ausencia de autoanticuerpos y de alteraciones metabólicas óseas, y la presencia de fenómenos de remodelación en el estudio radiológico, hacen pensar en una enfermedad de Gaucher. El diagnóstico diferencial con una enfermedad neoplásica puede ser más difícil, si bien la biopsia ósea permitiría observar los típicos macrófagos cargados de lípidos (células de Gaucher).
Un paciente con enfermedad de Fabry puede acudir a la consulta refiriendo acroparestesias y bro-tes de dolor osteoarticular intenso con escasos hallazgos en la exploración física. Probablemente se piensa inicialmente en un síndrome del túnel carpiano, en una polineuritis o en el inicio de una enfermedad inflamatoria articular de mecanismo inmunitario. Se trata de un conjunto de manifestaciones clínicas muy frecuentes en una consulta de reumatología. Si además hay proteinuria, insuficiencia renal y alteraciones vasculares o neurológicas, se puede plantear el diagnóstico de lupus, de vasculitis o de amiloidosis primaria. Además, el desarrollo de la enfermedad vascular y de la insuficiencia renal suele ser de aparición tardía, lo que dificulta el diagnóstico en las fases iniciales. En estos pacientes, la aparición de angioqueratomas o de opacidades corneales puede ser la clave del diagnóstico.
La enfermedad de Niemann-Pick tipo A (neuropática) comienza en etapas tempranas de la vida con hepatoesplenomegalia y afectación neurológica grave, y habitualmente se diagnostica en una consulta de pediatría. Sin embargo, la forma de Niemann-Pick no neuropática puede comenzar entre la primera y la segunda décadas de la vida con hepatoesplenomegalia y manifestaciones musculoesqueléticas, con dolor óseo, retraso en la madu ración esquelética, osteopenia radiológica y ensanchamiento del espacio medular con trabeculación grosera y alteración del contorno óseo con deformidad de Erlenmeyer similar a la enfermedad de Gaucher. El diagnóstico diferencial de un paciente en las primeras décadas de la vida con dolor óseo, anemia y osteopenia con alteración trabecular se puede plantear con una leucemia y también con una talasemia, cuyas alteraciones radiológicas son muy similares. Asimismo, el reumatólogo puede plantearse la posibilidad de que se trate de una artritis idiopática juvenil de comienzo. La ausencia de alteraciones en la fórmula leucocitaria, la ausencia de hemólisis y de los parámetros típicos de la artritis idiopática juvenil (elevación de reactantes de fase aguda, factor reumatoide, anticuerpos antinucleares) y de artritis en la exploración física descartan estos diagnósticos.
Los pacientes con enfermedad de Pompe con la forma de comienzo del adulto pueden acudir a la consulta con hipotonía y pérdida de fuerza progresiva, con afectación predominante proximal y elevación de las enzimas musculares. El diagnóstico inicial puede ser de miositis de mecanismo inmunitario, si bien obliga a valorar un amplio diagnóstico diferencial con las distrofias musculares, las miopatías inducidas por fármacos y otras miopa-tías metabólicas. El estudio electrofisiológico con descargas miopáticas y la biopsia muscular con una miopatía vacuolar puede orientar al diagnóstico, que se confirma por la determinación de glucosidasa a ácida en los leucocitos o los fibroblastos (biopsia de piel).
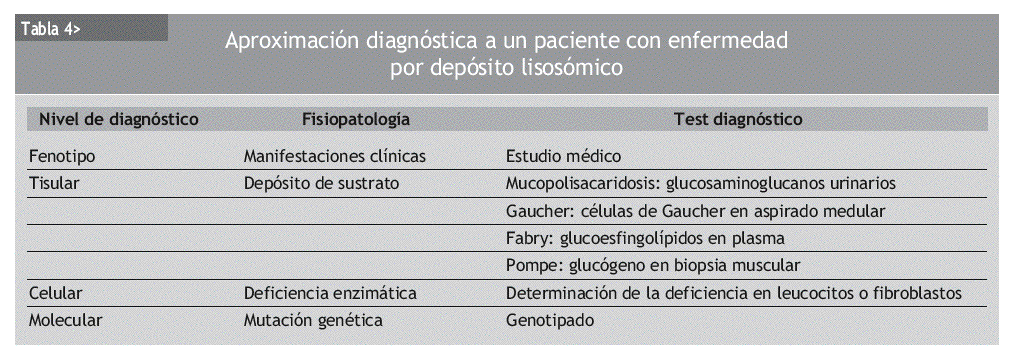
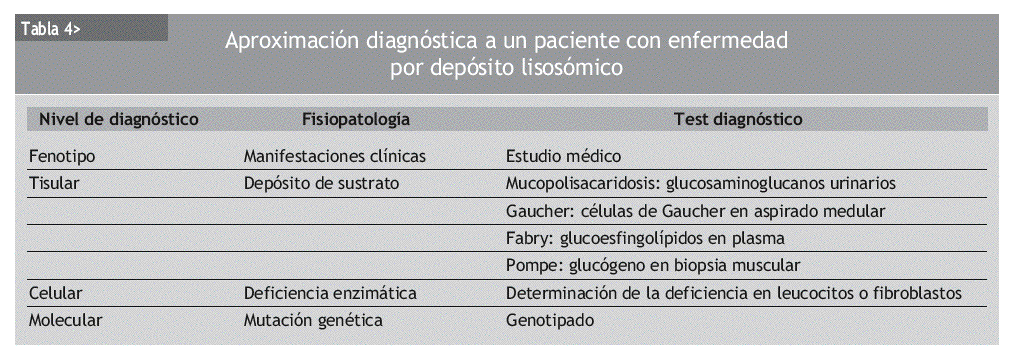
La tabla 4 muestra los diferentes niveles de diagnóstico ante la posibilidad de encontrarnos frente a una enfermedad por depósito lisosómico. En un primer nivel, las características clínicas del paciente orientan al diagnóstico. Posteriormente es posible identificar la sustancia que se está depositando, y finalmente determinar la deficiencia enzimática. El genotipado debe realizarse tanto al paciente como a la familia.
El conocimiento de las características fundamentales de las MPS, las lipidosis o las glucogenosis permite orientar el diagnóstico hacia estas enfermedades. Las nuevas técnicas diagnósticas son accesibles, rápidas y sencillas. El desarrollo durante los últimos años del tratamiento enzimático sustitutivo de especial relevancia al diagnóstico precoz de estos pacientes, mejorando el pronóstico de forma significativa y evitando en muchos casos un desenlace fatal.
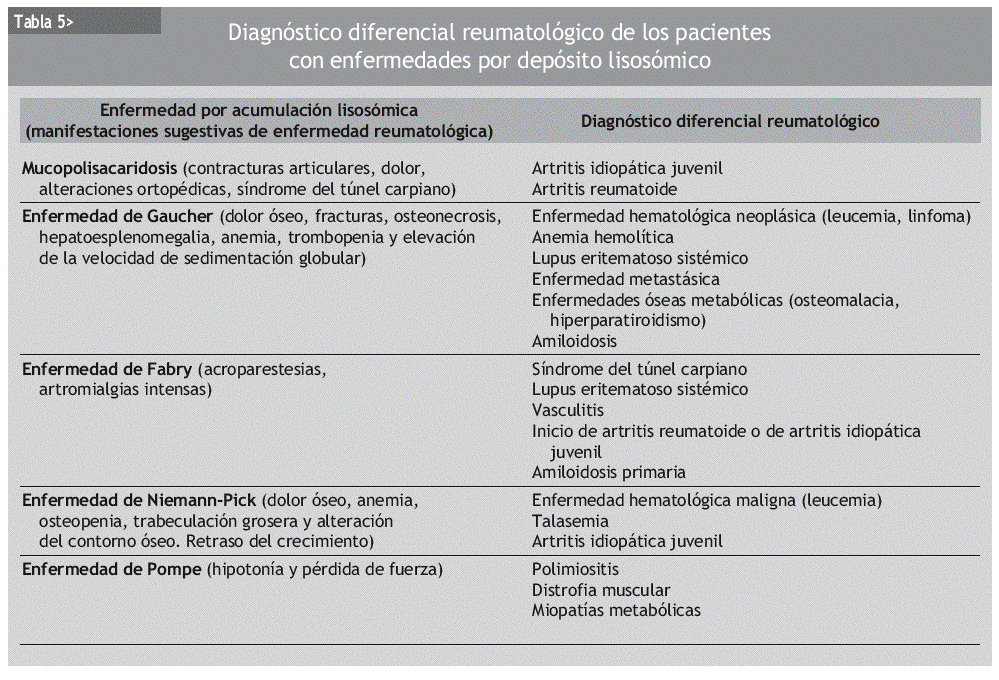
Las enfermedades por depósito lisosómico cursan con manifestaciones osteoarticulares (tablas 3 y 5), a menudo en los estadios más tempranos. Estos pacientes pueden ser valorados por reumatólogos, que de esta forma tienen la posibilidad de diagnosticar la enfermedad en sus fases más tempranas, optimizando así las posibilidades de que un eventual tratamiento sea más efectivo.
Agradecimientos
Queremos expresar nuestro agradecimiento al Dr. Sanjurjo, del Servicio de Pediatría del Hospital de Cruces, y a Francisco Abad, de Laboratorios Genzyme, que nos facilitaron valiosa información y nos ayudaron en la redacción de este trabajo.