



El síndrome de Cogan (SC) se caracteriza por la asociación de queratitis intersticial no luética y síntomas audiovestibulares (similares al síndrome de Ménière). Puede presentarse de forma atípica con manifestaciones oculares y audiovestibulares variadas, así como con manifestaciones sistémicas asociadas. Su nombre deriva del autor que la describió por primera vez. Afecta a adultos de ambos géneros, con edad media de 30años. La prevalencia está aumentada en la etnia caucásica. La etiopatogenia de este síndrome es desconocida, pero probablemente sea el resultado de un mecanismo autoinmune desencadenado por una infección. El diagnóstico es fundamentalmente clínico, mediante los criterios establecidos por Haynes et al. en 1980 para «SC típico» y «SC atípico». Es necesario establecer el diagnóstico diferencial con otras enfermedades sistémicas que cursan con manifestaciones similares en ojos y oído interno. El curso es variable, siendo la cofosis una complicación frecuente. El tratamiento precoz y mantenido es la base de una buena evolución.
Cogan syndrome (CS) is typified by nonsyphilitic interstitial keratitis and Meniere-like auditory involvement. It can present atypically with other ocular and audiovestibular symptoms and associated systemic manifestations. Its name derives from the author who first described the disease. CS affects adults of both sexes, with a mean age of 30years. The prevalence is higher in Caucasians. The pathogenesis of this syndrome is unknown, but is probably the result of an autoimmune mechanism triggered by an infection. The diagnosis is mainly clinical, using the criteria established by Haynes et al. in 1980 for “typical CS” and “atypical CS”. A differential diagnosis should be performed with other systemic diseases that cause similar eye and inner ear manifestations. The course is variable and deafness is a common complication. Prompt treatment and its maintenance are the basis of a favorable outcome.
El síndrome de Cogan (SC) es una enfermedad poco frecuente, de origen desconocido y caracterizada por inflamación ocular y síntomas audiovestibulares. Puede asociarse a manifestaciones sistémicas, tales como aortitis y vasculitis1.
Se conoce desde hace más de 60años2. La primera descripción en la literatura médica fue en 1945, y la realizó el oftalmólogo estadounidense David G. Cogan3. Posteriormente, en 1980, Haynes et al.4 sugirieron que este síndrome podía incluir otros síntomas oculares diferentes a la clásica queratitis intersticial y síntomas audiovestibulares variados, proponiendo criterios diagnósticos para el «SC típico» y «SC atípico».
Debido a la baja incidencia de esta enfermedad y a la ausencia de herramientas diagnósticas específicas, el diagnóstico es a menudo difícil, lo que puede condicionar retraso en su detección y agravar el pronóstico funcional, incluyendo riesgo de cofosis a largo plazo.
EpidemiologíaAfecta predominantemente a adultos jóvenes, con una edad media de inicio de los síntomas a los 30años. Se han descrito casos en edades pediátricas, a partir de los 4años5, y formas tardías, más allá de los 60años6. No existe un predominio de género.
La incidencia y la prevalencia no son bien conocidas, pero el número de casos según la literatura se estima en torno a 3001. No se ha descrito predominio étnico; sin embargo, no existen series asiáticas, por lo que parece afectar esencialmente a sujetos caucásicos.
EtiopatogeniaLos mecanismos responsables de la afectación ocular y del oído interno son desconocidos. Se sugiere que existe una reactividad autoinmune mediada por células contra antígenos de la córnea o componentes del oído interno7. Estos antígenos presentan homología con autoantígenos Ro/SSA y proteína lambda1 del core de reovirusiii, entre otros agentes infecciosos. En los animales, la transferencia pasiva de estos autoantígenos reproduce características del SC, lo que apoya la hipótesis de que la infección puede estar implicada en el desencadenamiento de esta enfermedad a través del mimetismo molecular7. Esto se ve apoyado por las observaciones de que hasta el 25% de los pacientes presentan un síndrome prodrómico de tipo viral8. No obstante ninguno de los estudios realizados para identificar un agente infeccioso (Chlamydia sp., Borrelia burgdorferi, citomegalovirus, virus de hepatitis B y C) lo ha demostrado9.
Se han aislado también en el suero de estos pacientes anticuerpos contra un antígeno peptídico («péptido Cogan»), que comparte homología con CD148 y conexina26, expresados en las células endoteliales y en el oído interno. La presencia de estos antígenos endoteliales añade evidencia adicional para la naturaleza autoinmune de esta enfermedad9.
Los anticuerpos anticitoplasma de neutrófilos específicos para mieloperoxidasa (pANCA) se han aislado hasta la fecha en 5 casos de SC10. El factor reumatoide, los anticuerpos antinucleares y una disminución de los niveles de complemento se han detectado en una minoría de estos pacientes. Se sugiere que los anticuerpos anti-Hsp70 pueden ser un marcador de pérdida de audición de origen autoinmune, pero no hay datos en la literatura que puedan concluir su utilidad para fines de diagnóstico o para establecer correlaciones con la actividad de la enfermedad10. En la serie de la Clínica Mayo se investigó en 10pacientes y fueron negativos en el 90% de los casos6.
El examen histopatológico de hueso temporal suele revelar infiltración de células plasmáticas y linfocitos del ligamento espiral, hidropesía endolinfática, cambios degenerativos en el órgano de Corti, neoformación ósea extensa en el oído interno y desmielinización y atrofia de las ramas vestibular y coclear del octavo par craneal. El estudio del tejido corneal evidencia infiltración de células linfocíticas y plasmáticas en las capas más profundas. A pesar de la asociación con vasculitis sistémica, los estudios de tejido corneal o de oído interno no revelan evidencia de vasculitis. En estos casos, la pared de los vasos afectados fuera del ojo o del oído interno mostrarán cambios histopatológicos típicos de inflamación aguda o crónica dependiendo de la naturaleza de la vasculitis (por lo general similares a la enfermedad de Takayasu o a la poliangeítis). Así mismo, en casos de afectación de tejido valvular el examen microscópico aprecia infiltración linfoide celular, necrosis fibrinoide y degeneración mixoide11.
Manifestaciones clínicasLa afectación característica ocular y/o del oído interno es necesaria para establecer el diagnóstico. Menos del 5% de los pacientes se presentan inicialmente con manifestaciones sistémicas. En estos casos el diagnóstico solo puede establecerse después del desarrollo de enfermedad ocular o del oído interno8.
OcularesLas manifestaciones oftalmológicas son generalmente los síntomas iniciales de la enfermedad. El cuadro más característico es una queratitis intersticial de inicio brusco que generalmente causa hiperemia conjuntival, dolor, lagrimeo, fotofobia, sensación de cuerpo extraño y visión borrosa. La afectación suele ser bilateral, sobre todo con la evolución de la enfermedad6. Rara vez es asintomática.
El examen con lámpara de hendidura comúnmente demuestra un infiltrado corneal irregular, profundo y granular, en particular en su parte posterior. Las anomalías observadas son similares a las de las queratitis infecciosas, lo que puede retrasar el diagnóstico. En raras ocasiones la inflamación corneal no controlada puede conducir a la neovascularización y a la opacidad de la córnea, desencadenando la pérdida permanente de la visión12.
Se han descrito muchas otras manifestaciones oftalmológicas, aisladas o asociadas con queratitis intersticial. Entre ellas, uveítis anterior y posterior, epiescleritis, conjuntivitis, queratitis ulcerativa periférica, ulceración de la córnea, hemorragia conjuntival y subconjuntival, exoftalmos, papiledema, oclusión de la arteria retiniana, hemorragia retiniana, vasculitis retiniana, edema macular quístico, parálisis oculomotora y glaucoma1,13. La vasculitis retiniana y la escleritis posterior son enfermedades graves que con frecuencia causan discapacidad visual y exigen intervención terapéutica urgente.
AudiovestibularesLa afectación audiovestibular puede ser la primera manifestación o, más frecuentemente, ser posterior a las manifestaciones oftalmológicas con un intervalo libre de meses o incluso años. Normalmente se presenta como un síndrome de Ménière con aparición repentina de vértigo, tinnitus, náuseas o vómitos, nistagmo, ataxia y disminución de la audición. Sin embargo, en el SC el cuadro clínico es más grave, debilitante, prolongado y generalmente bilateral6.
La pérdida auditiva es constante durante la evolución de la enfermedad, siendo típicamente fluctuante y progresiva, lo que conduce a la sordera de percepción uni o bilateral. Se han descrito casos sin manifestaciones vertiginosas asociadas.
Los episodios recurrentes de afectación del oído interno pueden resultar en hidropesía coclear, condición que se asocia a fluctuaciones en la audición debido a los cambios en la presión coclear independientes de la inflamación. Estos episodios suelen ser difíciles de distinguir de los de origen inflamatorio. La ausencia de otros datos de actividad (inflamación ocular, manifestaciones sistémicas) o la presencia de infección respiratoria superior o menstruación como desencadenante debe hacernos sospechar del origen no inflamatorio.
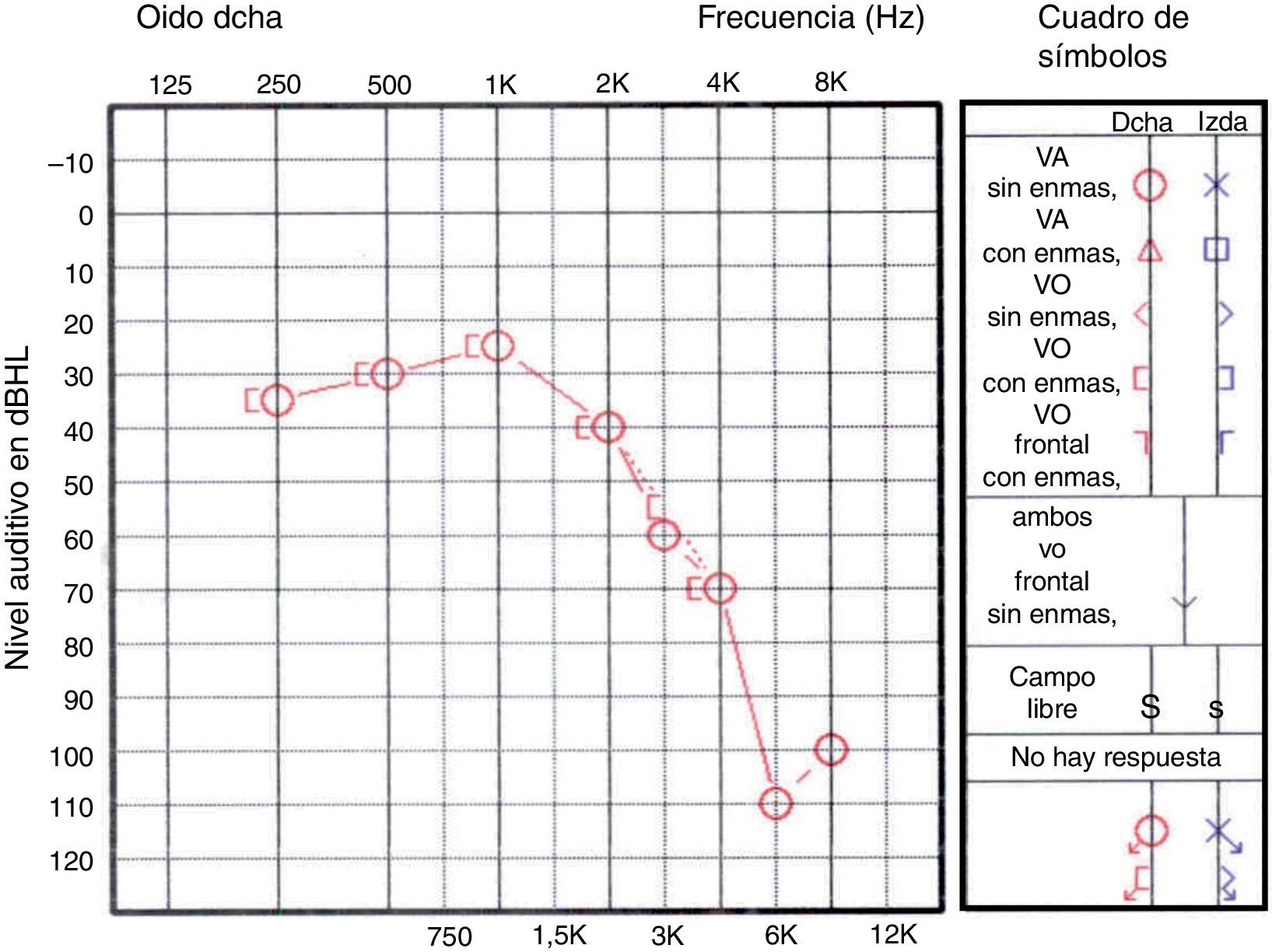
La audiometría muestra una hipoacusia neurosensorial con afectación predominante de altas y bajas frecuencias (fig. 1), asociada generalmente a una disminución de la capacidad de discriminación de las palabras14. Los potenciales evocados auditivos confirman el daño coclear. La evaluación de la función vestibular (prueba calórica) evidencia anomalías bilaterales en el 93% de los casos.
Sistémicas
Las vasculitis sistémicas se desarrollan en el 15-20% de los casos, por lo general varios años después del diagnóstico15. Las vasculitis de mediano y gran vaso son las más frecuentes, aunque pueden afectarse también vasos de pequeño tamaño. Lo más característico es la participación del arco aórtico, con posibles aneurismas y estenosis, pudiendo ocurrir también la disección aórtica16. La aortitis ha sido descrita en el 10% de los pacientes. Puede causar dilatación proximal de la aorta, insuficiencia valvular aórtica, enfermedad de la arteria coronaria ostial y aneurismas aórticos toracoabdominales. La afectación inflamatoria del tejido de conducción cardíaco puede ser responsable de bloqueos auriculoventriculares.
Se han descrito accidentes isquémicos cerebrovasculares por afectación de la arteria carótida interna o el territorio vertebrobasilar. El tracto gastrointestinal puede estar afectado por arteritis mesentérica, responsable de diarreas, hemorragias digestivas y dolor abdominal. La afectación renal secundaria a estenosis de la arteria renal o glomerulonefritis también puede ocurrir.
Algunos casos asociados a ANCA (de tipo pANCA) plantean problemas potenciales de los posibles vínculos entre vasculitis asociadas a ANCA y el SC. Por otra parte, se ha descrito asociación de este síndrome con enfermedades inflamatorias crónicas del intestino (enfermedad de Crohn y colitis ulcerosa)17. Recientemente un SC atípico fue diagnosticado en un paciente con fiebre mediterránea familiar genéticamente confirmado18.
Los síntomas generales (fiebre, fatiga, pérdida de peso) están presentes en casi la mitad de los casos. Aproximadamente un tercio de los pacientes presentan manifestaciones articulares tales como mialgia, artralgia y a veces artritis. Otras manifestaciones sistémicas inespecíficas descritas en algunos casos son: linfadenopatía, fenómeno de Raynaud, hepatoesplenomegalia, pleuritis, condritis, mononeuropatía múltiple, encefalopatía, meningitis aséptica, epilepsia, parálisis facial, mielopatía, dolor testicular, erupciones en la piel, úlceras bucales y pioderma gangrenoso.
Destacar que a diferencia del SC típico, el atípico se asocia más comúnmente a manifestaciones sistémicas19 y a otras enfermedades autoinmunes, tales como sarcoidosis, artritis reumatoide, policondritis recidivante, artritis idiopática juvenil, síndrome de Sjögren y enfermedad inflamatoria intestinal, entre otras. Esto podría explicar la dificultad que suele entrañar su diagnóstico y la mayor resistencia al tratamiento inmunosupresor.
DiagnósticoNo existen criterios diagnósticos ni de clasificación validados20. El diagnóstico se basa en la presencia de enfermedad ocular inflamatoria característica y disfunción vestibuloauditiva. La evaluación inicial debe constar de un examen oftalmológico, neurológico y otológico, así como sistémico, para buscar evidencia de vasculitis. Desde que en 1980 Haynes et al.4 sugirieran los criterios diagnósticos de síndrome de Cogan típico y atípico (tabla 1), esta clasificación se ha utilizado durante muchos años. No obstante, a veces puede resultar confusa, ya que un mismo paciente puede llegar a cumplir criterios para ambos patrones6. Existe una tendencia actual a abandonar los términos SC «típico» y «atípico» porque parecen no influir en el pronóstico, aunque se requiere de más estudios para poder confirmar esta hipótesis21.
Criterios diagnósticos de síndrome de Cogan
| Síndrome de Cogan típico cuando cumple estas 3 condiciones: |
| a) Manifestaciones oculares típicas de queratitis intersticial no sifílica, eventualmente asociada a conjuntivitis, iritis, hemorragia conjuntival o subconjuntival |
| b) Manifestaciones audiovestibulares similares al síndrome de Ménière (inicio brusco con náuseas, vómitos, vértigo y acufenos) con sordera progresiva en 1 o 2 meses |
| c) Intervalo entre la aparición de manifestaciones oculares y audiovestibulares inferior a 2 años |
| Síndrome de Cogan atípico: comprende 3 situaciones posibles |
| a) Manifestaciones oftalmológicas atípicas (epiescleritis, escleritis, coroiditis, oclusión arterial retiniana, hemorragia retiniana, edema papilar, exoftalmos) asociadas a manifestaciones audiovestibulares típicas ménièriformes con un intervalo entre ellas inferior a 2 años |
| b) Manifestaciones oftalmológicas típicas asociadas a manifestaciones audiovestibulares diferentes del síndrome de Ménière con un intervalo entre ellas inferior a 2 años |
| c) Manifestaciones oftalmológicas y audiovestibulares típicas con un intervalo entre ellas superior a 2 años |
Fuente: Haynes et al.4.
Los estudios de laboratorio suelen revelar un síndrome inflamatorio, con aumento de reactantes de fase aguda y, a veces, leucocitosis con neutrofilia. El estudio de anticuerpos antinucleares (ANA), ANCA, anticuerpos antifosfolípidos y factor reumatoide suele ser negativo, y el complemento, normal. No obstante, como se ha indicado anteriormente, a veces pueden ser positivos los estudios de autoinmunidad10.
Algunos autores consideran que la RM cerebral puede ser útil para el diagnóstico, pudiendo mostrar estrechamiento u obliteración del laberinto vestibular así como hallazgos compatibles con señales inflamatorias22. Pero en la mayoría de las series publicadas los resultados han sido negativos, siendo útil principalmente para descartar tumores o lesiones isquémicas, incluyendo el ángulo pontocerebeloso.
La tomografía por emisión de positrones (PET) con 2-desoxi-2-[18F] glucosa-D-fluoro (FDG) está siendo cada vez más importante en el diagnóstico, la estratificación, el tratamiento y el seguimiento en oncología clínica y se ha utilizado recientemente en el diagnóstico de enfermedades infecciosas con elevado metabolismo de glucosa intracelular. Las células inflamatorias activadas sobreexpresan transportadores de glucosa y acumulan mayores cantidades de esta sustancia y otras estructuralmente relacionadas, tales como F18-FDG. Por lo tanto, FDG-PET también se introduce como medio de diagnóstico para evaluar la participación de grandes vasos en las vasculitis, correlacionándose positivamente la captación con los marcadores de fase aguda. Otras modalidades diagnósticas estándar como la biopsia, la angiografía, la ecografía y la RM son habitualmente incapaces de demostrar el alcance total de la afectación vascular. En la PET/TC, el componente tomografía computarizada (TC) es útil en la localización precisa de la anomalías y puede proporcionar información sobre los cambios en la estructura de la pared o flujo luminal23. Dado el mal pronóstico asociado con la participación de los grandes vasos, sería interesante ver si la PET/TC podría ser utilizada en personas con enfermedad temprana para identificar vasculitis subclínicas y, por tanto, la necesidad de regímenes inmunosupresores agresivos. Del mismo modo, ese enfoque podría ser útil en el seguimiento de pacientes con aortitis para documentar respuesta al tratamiento. Se requieren estudios adicionales en esta área para determinar si esa modalidad de imagen puede ser un complemento útil en el diagnóstico y en la estratificación del riesgo en pacientes con SC.
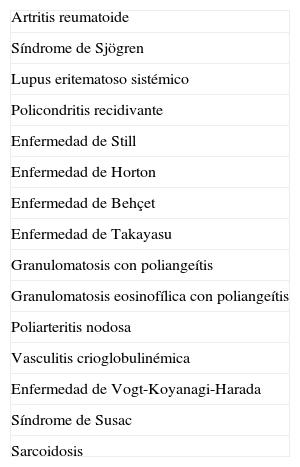
Diagnóstico diferencialEl diagnóstico diferencial del SC debe incluir otras enfermedades sistémicas que causen manifestaciones similares en ojos y oído interno (tabla 2).
Diagnóstico diferencial en el síndrome de Cogan
| Artritis reumatoide |
| Síndrome de Sjögren |
| Lupus eritematoso sistémico |
| Policondritis recidivante |
| Enfermedad de Still |
| Enfermedad de Horton |
| Enfermedad de Behçet |
| Enfermedad de Takayasu |
| Granulomatosis con poliangeítis |
| Granulomatosis eosinofílica con poliangeítis |
| Poliarteritis nodosa |
| Vasculitis crioglobulinémica |
| Enfermedad de Vogt-Koyanagi-Harada |
| Síndrome de Susac |
| Sarcoidosis |
Otras posibilidades diagnósticas son las causas infecciosas de queratitis intersticial, como la sífilis congénita, la tuberculosis, la infección por Chlamydia y la infección viral. Los agentes infecciosos pueden ser diferenciados de SC por pruebas serológicas (p.ej., FTA-Abs para la sífilis), pruebas cutáneas (p.ej., prueba de la tuberculina), cultivos, ensayos terapéuticos con tratamiento antimicrobiano (tetraciclinas para Chlamydia), y los signos clínicos característicos (p.ej., patrón dendrítico en infección corneal herpética).
El SC también debe diferenciarse de un síndrome de encefalopatía subaguda, pérdida auditiva neurosensorial aislada y oclusiones de las arteriolas retinianas en adultos jóvenes por vasculopatía oclusiva no vasculítica. Los principales hallazgos de estas enfermedades, que nos permite diferenciarlos del SC, incluyen la presencia de alteraciones neuropsiquiátricas, la ausencia de queratitis intersticial u otra inflamación en segmentos anteriores del ojo, así como alteraciones retinianas características en la exploración oftalmológica.
PronósticoEl curso del SC es variable15. Por lo general, después de un brote inicial de varias semanas o meses evoluciona de forma lenta y progresa hacia la cronicidad. No obstante, puede cursar en forma de brotes recurrentes de afectación ocular y/o vestibular, a intervalos variables.
Generalmente el pronóstico visual es bueno. Una disminución transitoria y moderada de la agudeza visual es habitual, siendo raros los casos de ceguera. La sordera es una complicación frecuente, desarrollándose hasta en el 50% de los pacientes. En base a esto, se recomienda un seguimiento estrecho con audiogramas mensuales durante los 3 primeros meses, cada 3meses durante los 2 primeros años, cada 6meses el tercer año y una vez al año posteriormente. Esto nos permitirá la detección precoz de complicaciones potenciales y la instauración de las medidas terapéuticas más adecuadas.
Los pacientes sin enfermedad sistémica generalmente tienen buen pronóstico y buena expectativa de vida. Por el contrario, cuando se desarrolla vasculitis grave existe mayor riesgo de muerte debido a complicaciones19. Se han informado varias muertes debidas a insuficiencia cardíaca, infarto de miocardio, accidente cerebrovascular y hemorragia subaracnoidea. Por otra parte, la morbilidad suele ser alta debido a la incapacidad impuesta por el deterioro auditivo y por los efectos adversos de los agentes inmunosupresores necesarios en la mayoría de los casos.
TratamientoEl tratamiento del SC no está bien establecido. La información disponible en la literatura se basa en informes de casos clínicos, ya que no se dispone de ensayos aleatorizados que comparen las diferentes opciones terapéuticas19. Ninguno es totalmente eficaz, y el tratamiento con corticoides es el más usado.
Manifestaciones oftalmológicasLa queratitis intersticial responde bien a esteroides tópicos, con excelente pronóstico funcional si se inicia el tratamiento de forma precoz9. Suele asociarse al tratamiento colirios de atropina por su efecto midriático y excepcionalmente son necesarias otras opciones terapéuticas locales, como la ciclosporina. La queratitis intersticial justifica raramente tratamiento sistémico. Por el contrario, otras manifestaciones oftalmológicas (escleritis, queratitis ulcerativa) a menudo requieren glucocorticoides orales (1mg/kg/día). La falta de respuesta en 2-3semanas, la incapacidad de disminuir la dosis de corticoides a 10mg/día de prednisona o el desarrollo de toxicidad inducida por glucocorticoides son indicaciones para el uso de inmunosupresores4. Los más utilizados son metotrexato (15-25mg/semana), ciclofosfamida oral (2-3mg/kg/día) y ciclosporina (5mg/kg/día).
La opacidad corneal progresiva puede requerir un trasplante de córnea. Así mismo, puede ser necesaria una intervención quirúrgica para corregir las cataratas secundarias a corticoterapia. El umbral para estos procedimientos es menor en pacientes con discapacidad auditiva asociada, como ocurre en este síndrome, ya que el lenguaje de signos puede ser fundamental para la comunicación24.
Manifestaciones audiovestibularesEl compromiso de la agudeza auditiva justifica la corticoterapia sistémica. El tratamiento debe iniciarse de forma precoz, ya que la falta de control de un brote puede conducir a la sordera profunda definitiva1.
En casos graves, como pérdida de agudeza auditiva importante y/o manifestaciones sistémicas graves asociadas, la administración inicial puede ser en forma de bolos de metilprednisolona de 500-1.000mg/día durante 3días consecutivos. Luego se continúa con la dosis habitual de 1mg/kg/día de prednisona oral, reduciendo la dosis de forma muy gradual, disminuyendo aproximadamente 5 a 10mg cada 1 o 2semanas, hasta completar 4 a 6meses de tratamiento. En caso de recaídas, se recomiendan glucocorticoides de mantenimiento (5-10mg/día), así como tratamiento ahorrador de los mismos. Los glucocorticoides intratimpánicos pueden ser eficaces en algunos pacientes con pérdida auditiva neurosensorial idiopática25, por lo que puede considerarse su uso en este síndrome, aunque su eficacia no ha sido demostrada.
La evaluación audiométrica se lleva a cabo a las 2 a 4semanas del inicio de los glucocorticoides. Si existe mejoría objetiva y subjetiva se mantiene en el régimen anterior. Por el contrario, en ausencia de la mejoría auditiva, los glucocorticoides deben reducirse rápidamente y después suspenderse (a menos que sean necesarios para el control de las manifestaciones oculares o sistémicas de la enfermedad), ya que la recuperación de la audición raramente va a ocurrir transcurrido este período26.
Dado que estos pacientes pueden presentar pérdida auditiva por hidropesía coclear, debe plantearse el tratamiento con diuréticos. La hidroclorotiazida a dosis de 25mg/día y la furosemida a dosis de 10-20mg/día han sido los más utilizados. No obstante, ante la ausencia de mejoría a los 4 a 7días está indicado tratamiento con glucocorticoides.
El uso de terapias inmunosupresoras debe considerarse en pacientes que requieren dosis de glucocorticoides muy altas para controlar la pérdida de audición o por toxicidad secundaria a estos fármacos. El metotrexato es actualmente el más utilizado, sobre todo como ahorrador de esteroides (dosis de hasta 25mg/semana)27. En segundo lugar el más utilizado es la azatioprina (dosis de 1,5 a 2,5mg/kg/día). Cuando los anteriores fracasan, la eficacia de otros inmunosupresores (ciclosporina, leflunomida, micofenolato de mofetilo) es difícil de evaluar, basándose su uso en varios casos clínicos publicados6,19. La ciclofosfamida intravenosa se ha utilizado para manifestaciones oculares graves1, y su eficacia en los síntomas audiovestibulares es variable28.
Recientemente se está investigando la eficacia de los fármacos biológicos. Con respecto a los antagonistas del TNF-α9, el etanercept no ha evidenciado ser eficaz en la preservación o mejoría de la pérdida auditiva; sin embargo, mejora la capacidad de discriminación. Por el contrario, el infliximab parece ser eficaz en la inducción y mantenimiento de la remisión en pacientes con SC resistentes a glucocorticoides e inmunosupresores. Se sugiere que la eficacia de este fármaco será mayor si se inicia en una etapa temprana de la enfermedad, cuando las lesiones son aún reversibles, por lo que se plantea el infliximab como tratamiento de primera línea y no solo como rescate en enfermedad resistente, requiriéndose estudios controlados aleatorizados que confirmen dicha hipótesis28.
La eficacia del rituximab se ha descrito en un paciente. En este caso, la terapia convencional (glucocorticoides, metotrexato, ciclofosfamida y ciclosporina) consiguió buen control de las manifestaciones oftalmológicas y vestibulares, pero la persistencia del deterioro de la función auditiva obligó a usar un antagonista del TNF-α (adalimumab), sin éxito. Se decidió usar de forma compasiva rituximab, en dosis de 500mgi.v. una vez a la semana durante 4semanas consecutivas y se repitió a los 6meses. Los controles audiométricos posteriores mostraron mejoría de la audición y control de todas las manifestaciones clínicas. Por ello se postula al rituximab como agente importante para evitar la sordera y la necesidad de implantes cocleares en casos graves29.
El SC es considerado actualmente como una vasculitis, por lo que el tratamiento intensivo puede estar indicado con el fin de prevenir la progresión de la enfermedad y el daño irreversible vestibular, ocular o audiovestibular. En este sentido, se plantea la plasmaféresis como tratamiento eficaz, debiéndose considerar en pacientes con actividad inflamatoria crónica y resistente o efectos secundarios graves con otras opciones terapéuticas30.
Cuando la sordera es importante, en última instancia los pacientes pueden beneficiarse de implantes cocleares, con resultados funcionales satisfactorios inmediatos. Sin embargo, el destino a largo plazo de estos implantes no es bien conocido en este síndrome, ya que los test funcionales pueden degradarse debido a la osificación progresiva de la cóclea31.
Manifestaciones sistémicasEl tratamiento consiste en glucocorticoides que ocasionalmente se asocian a inmunosupresores. El recambio de la válvula aórtica a veces es necesario, así como la cirugía de los aneurismas torácicos y/o abdominales32.
ConclusionesEl SC afecta principalmente los ojos y el oído interno, pero en el 15-20% de los casos se asocia a vasculitis sistémicas implicando a múltiples órganos, entre los que se incluyen el corazón, el aparato digestivo y el riñón. Su etiopatogenia es desconocida, aunque se cree que hay una reacción de autoinmunidad contra antígenos de la córnea y del oído interno. El diagnóstico es clínico, cumpliendo una serie de criterios ya establecidos, pero la RM y la PET/TC pueden ser de gran ayuda tanto para el diagnóstico como para valorar posibles complicaciones. El tratamiento de elección son los glucocorticoides sistémicos, pero en casos resistentes se suele usar los inmunosupresores. El papel de los fármacos biológicos debe ser estudiado con más detalle. El pronóstico visual es generalmente bueno, mientras que la sordera es una complicación frecuente; siendo el implante coclear una alternativa terapéutica en estos casos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.