


Introducción
El síndrome de Stickler es un trastorno hereditario autosómico (la mayoría de las veces dominante) del tejido conjuntivo que pertenece a las llamadas colagenosis. Fue descrito en 1965 por Gunnar B. Stickler1 como una artro-oftalmopatía hereditaria progresiva1.
En la décima versión de la Clasificación Estadística Internacional de Enfermedades y otros Problemas de Salud (CIE-10)2 se ha clasificado con el código Q87.8: Otros síndromes de malformaciones congénitas especificados no clasificados en otra parte.
Se ha estimado que su incidencia aproximada es de 1 caso cada 10.000 nacimientos3. Se considera, sin que haya sido confirmado, la enfermedad congénita del colágeno más frecuente en Europa y América, con una prevalencia similar a la del síndrome de Marfan4.
Etiología y patogenia
El colágeno es una proteína fibrosa extracelular que forma parte del tejido conjuntivo. Es especialmente abundante en los tejidos que soportan peso, como los cartílagos y los huesos, y también en los tendones, las fascias y la dermis. Asimismo constituye el armazón de todos los órganos y vísceras del organismo.
Existen 40 genes diferentes que codifican como mínimo a 27 tipos diferentes de colágeno. En los ojos embrionarios y en los maduros se han encontrado 22 tipos de colágeno distintos, pero de éstos tan sólo 6 también están presentes en el cartílago articular: los colágenos 2, 5, 6, 9, 11 y 27. La mayoría de las fibras del colágeno en el cartílago y en el humor vítreo son heterotópicas, es decir, contienen más de un tipo de colágeno5.
El síndrome de Stickler se ha descrito a causa de la mutación muy heterogénea en 4 genes que controlan la síntesis del colágeno 2, 9 y 11, y por consiguiente es fenotípicamente muy variable en su expresión5-7.
El colágeno 2 es el que se halla en mayor proporción en el humor vítreo, en el cartílago y en los discos intervertebrales. El colágeno 9 se localiza asociado a fibrillas de colágeno tipo 2 en el cartílago articular maduro, en la córnea y en el humor vítreo. El colágeno 11 tiene una distribución similar a la del cartílago tipo 2. Los 3 tipos de colágeno se encuentran en la cóclea8,9.
Las mutaciones de los genes que actúan sobre el colágeno 2 y 11 son autosómicas dominantes, mientras que la que actúa sobre el colágeno 9 es autosómica recesiva10-13. Las más frecuentes son las autosómicas dominantes, por lo que todos los miembros de la familia de un paciente diagnosticado de síndrome de Stickler deben ser estudiados.
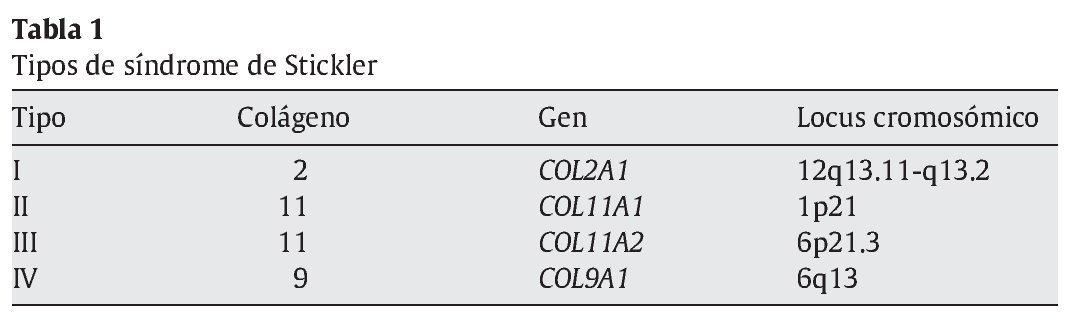
El síndrome de Stickler se ha clasificado en 4 tipos en dependencia del gen mutado (tabla 1), que asimismo se relacionan con el fenotipo oftalmológico y concretamente con las anormalidades en la arquitectura del gel vítreo14:
Síndrome de Stickler tipo I10 o fenotipo vítreo I (MIM 108300). Producido por la mutación en el gen COL2A1, es el más frecuente (aproximadamente el 75%) y causa un síndrome de Stickler completo. Cursa con manifestaciones oculares, craneofaciales, audiológicas y articulares. Desde el punto de vista oftalmológico se caracteriza por la persistencia de un gel residual inmediatamente detrás del cristalino y rodeado por una membrana festoneada.
Síndrome de Stickler tipo II11 o fenotipo vítreo II (MIM 604841). Producido por la mutación COL11A1, es mucho menos frecuente que el anterior y cursa con un síndrome de Stickler completo, si bien la artropatía no está necesariamente presente. Desde el punto de vista oftalmológico se caracteriza por presentar en el humor vítreo fibras que parecen abalorios.
Síndrome de Stickler tipo III12 (MIM 184840). Producido por la mutación COL11A2, causa un síndrome que afecta a las articulaciones y a los oídos sin afectar a los ojos. También se le conoce como displasia oto-espondilo-megaepifisaria.
Síndrome de Stickler tipo IV13. Se produce por la mutación COL9A1 y cursa con hipoacusia neurosensorial, miopía con vitreorretinopatía, y displasia epifisal. Desde el punto vista oftalmológico se caracteriza por una progresiva degeneración del humor vítreo que se manifiesta con una licuefacción de éste.
Hay otras entidades sin el fenotipo del síndrome de Stickler que están asociadas a mutaciones en los mismos genes15, como la acondrogénesis tipo 2 (Mendelian Inheritance in Man [MIM] 200610) por mutación en COL2A1, síndrome de Marshall (MIM 154780) por mutación en COL11A1, síndrome de Weissenbach-Zweymuller (MIM 277610) por mutación en COL11A2, y otros. Las mutaciones genéticas combinadas son infrecuentes.
Manifestaciones clínicas
Se pueden hallar signos muy variados4,14,16-19. Antes de los 10 años las consultas oftalmológicas son habituales, y a partir de la tercera década aumentan las consultas a consecuencia de los dolores articulares secundarios a los prematuros cambios degenerativos, más comunes en las rodillas y en las articulaciones coxofemorales.
Manifestaciones craneofaciales: cara plana muy frecuente con puente nasal ancho o bien plano y mejillas planas por hipoplasia malar (estas características faciales son más pronunciadas a edades tempranas); fhiltrum largo muy frecuente; retrognatia y o micrognatia; paladar hendido; paladar hendido submucoso; úvula bífida; paladar corto; maloclusión dental; erupción precoz de los dientes/dientes neonatales; anomalía del esmalte, y anodoncia/oligodoncia.
Manifestaciones oculares: cataratas precoces, incluso congénitas, muy frecuentes y no progresivas; miopía temprana muy frecuente (antes de los 6 años) generalmente alta, de -3 o más dioptrías, siendo habitual que supere las -6 e incluso las -8 dioptrías, habitualmente congénita y no progresiva; alto riesgo de desprendimiento de retina, que puede ocurrir en ambos ojos, normalmente antes de los 30 años e incluso en niños; pigmentación perivascular retiniana con adelgazamiento de la misma por degeneración; astigmatismo; buftalmia; subluxación del cristalino, que en algunos casos puede ser congénita y bilateral; glaucoma; estrabismo; epicanto; anomalía vítrea, y ceguera.
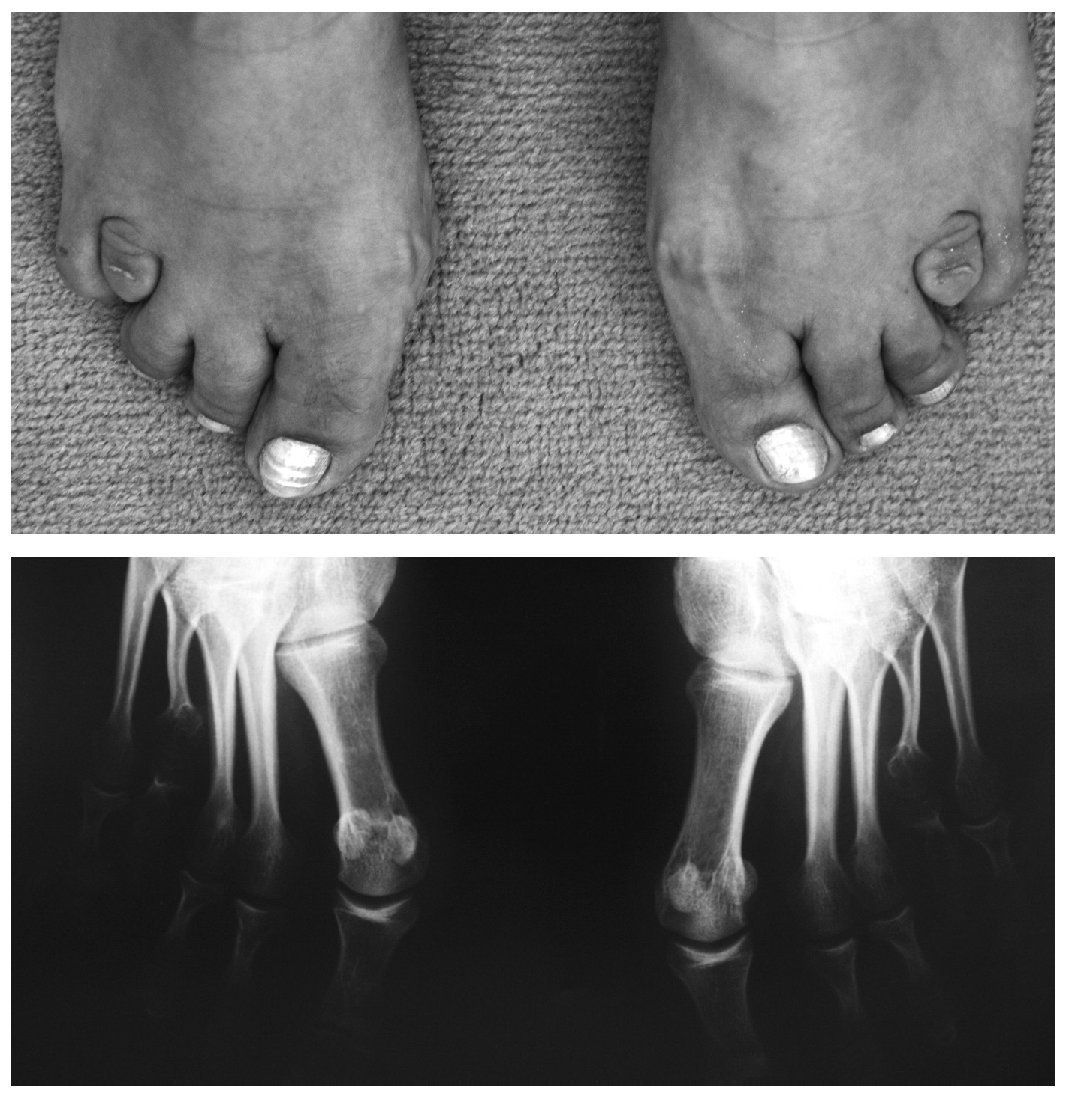
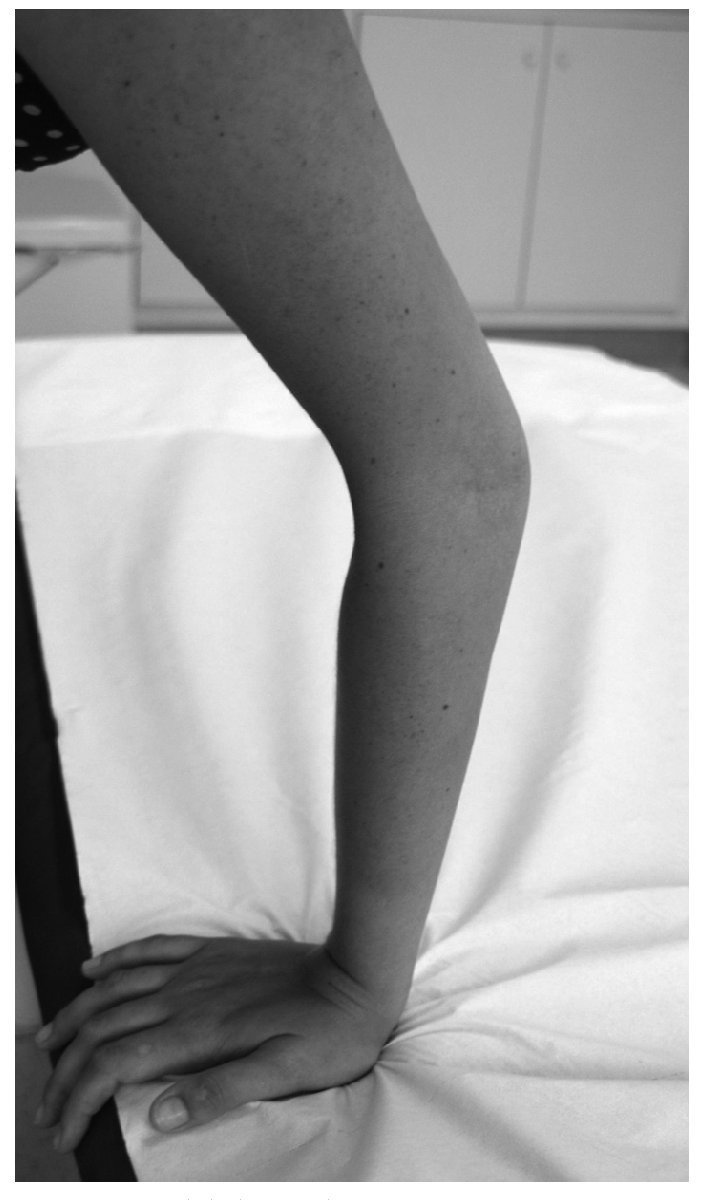
Manifestaciones osteomioarticulares: epífisis planas muy frecuentemente; ensanchamiento metafisario y epifisario; coxa valga frecuente, con ensanchamiento del cuello femoral; protrusión acetabular; epifisiólisis de la cabeza femoral; osteonecrosis avascular de la cabeza femoral; hipoplasia de los huesos pélvicos; genu valgum; acortamiento rizomélico; braquidactilia (fig. 1); escoliosis/cifosis; platiespondilia; placas terminales vertebrales irregulares (similar a la enfermedad de Scheuermann); espondilolistesis; hendidura coronal vertebral; aspecto marfanoide; adelgazamiento cortical relativo; osificación disarmónica de los huesos del carpo; pectus carinatum o excavatum; hipermovilidad articular que va disminuyendo con la edad (fig. 2); hipotonía; amiotrofia/agenesia muscular; artrosis de inicio precoz en la tercera o cuarta década, y crujidos articulares.
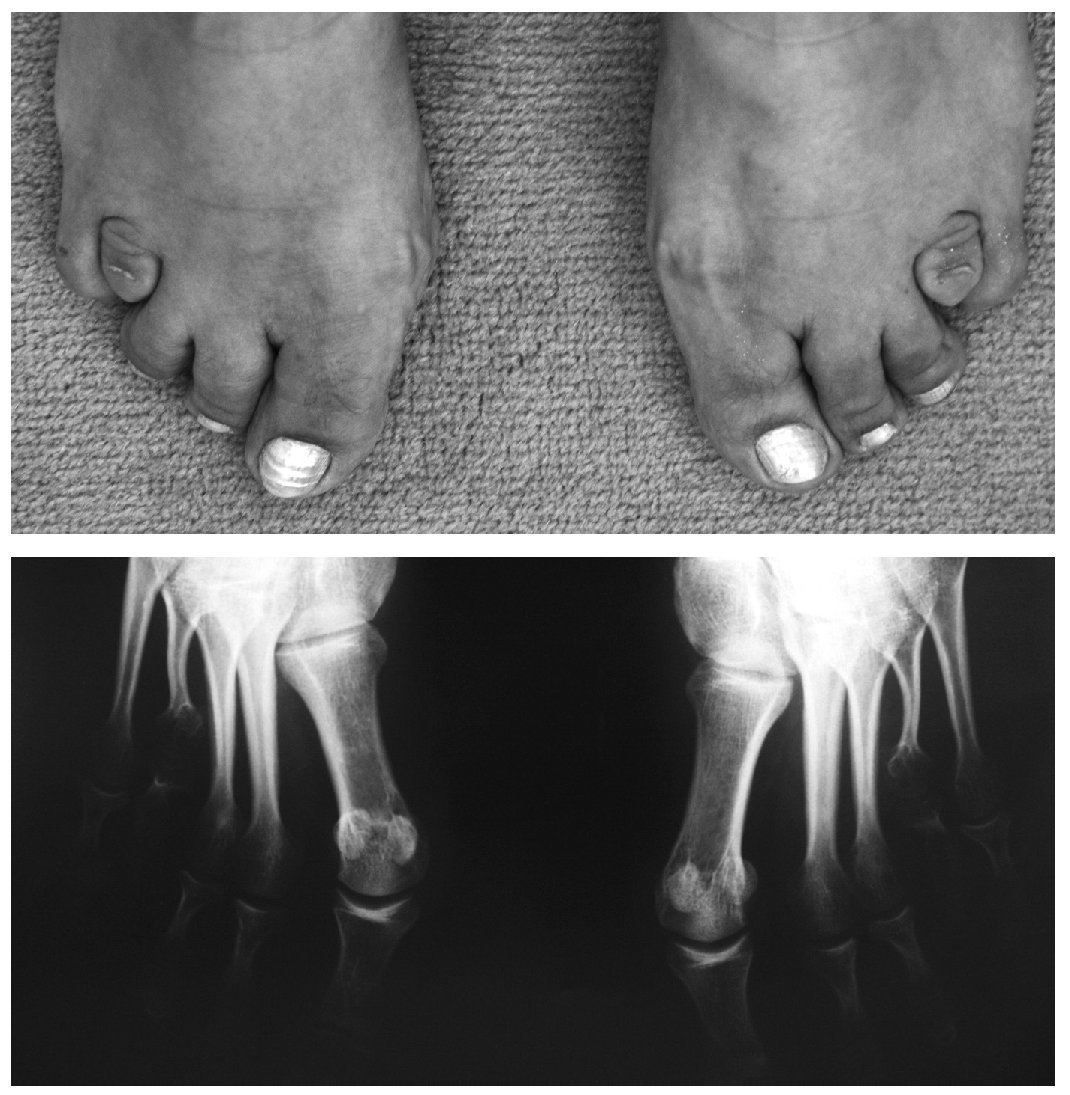
Figura 1. Braquidactilia bilateral.
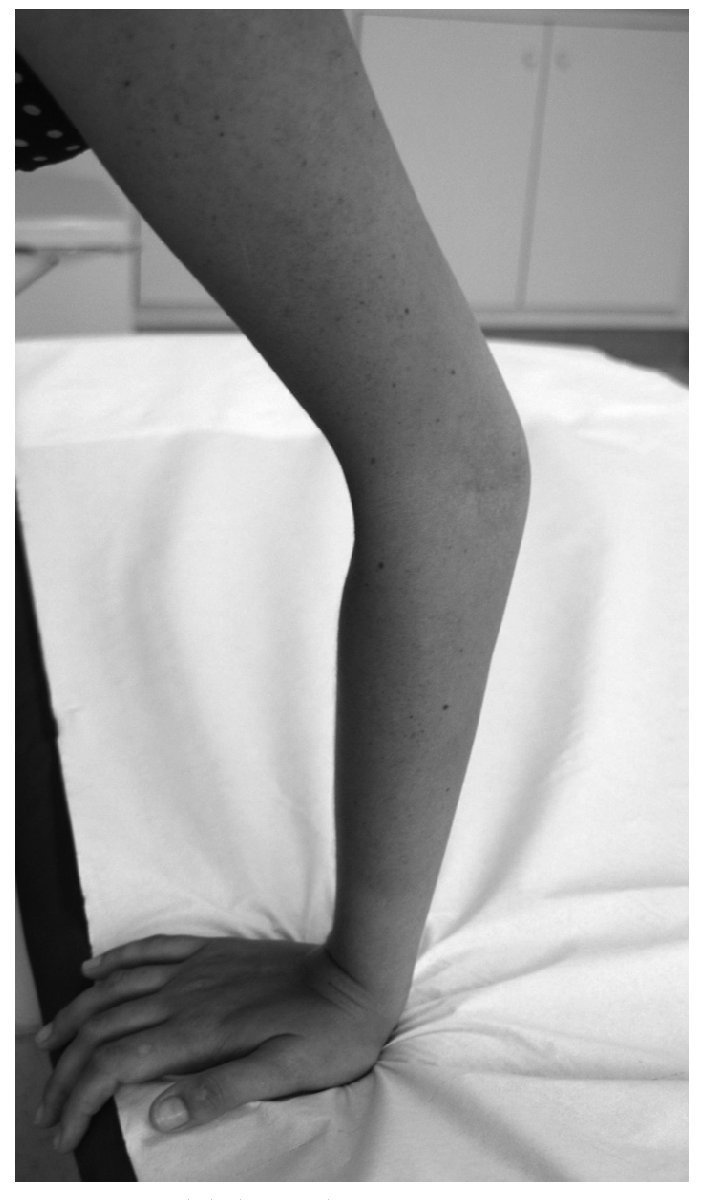
Figura 2. Hipermovilidad articular.
Manifestaciones otológicas: hipoacusia conductiva debido a disfunción de la trompa de Eustaquio secundaria al paladar hendido, o bien por anomalía en la cadena de huesecillos; hipoacusia neurosensorial progresiva que afecta a las frecuencias altas; hipoacusia mixta, y otitis media.
Otras manifestaciones: prolapso mitral (no confirmado en todas las series); retraso mental (infrecuente, y en todo caso moderado o ligero), y delgadez (salvo lipodistrofia).
Diagnóstico
Se ha sugerido que este diagnóstico debe considerarse en los individuos con hallazgos clínicos en 2 o más de estas categorías15:
Oftalmológicos:
- Cataratas congénitas o de inicio prematuro.
- Anomalías congénitas del vítreo, desprendimiento de retina o retinopatía con pigmentación paravascular reticular.
- Miopía superior a -3 dioptrías.
Nota: Los recién nacidos son típicamente hipermétropes (+1 dioptría o más); de este modo, el hallazgo de algún grado de miopía en un recién nacido con riesgo (p. ej., un recién nacido con la secuencia de Pierre-Robin [micrognatia, paladar duro hendido, glosoptosis] o un familiar con síndrome de Stickler) es sugestivo del diagnóstico de síndrome de Stickler).
Craneofaciales:
- Hipoplasia facial media, puente nasal deprimido, anteversión de las ventanas nasales (las características faciales son típicamente más pronunciadas en niños).
- Úvula bífida, paladar duro hendido.
- Micrognatia.
- Secuencia de Robin. El síndrome de Stickler es el diagnóstico más frecuente en pacientes con la secuencia de Robin20.
Audiológicos:
- Hipoacusia neurosensorial o de conducción.
- Oído medio con la cadena de huesecillos hipermóvil.
Articulares:
- Hipermovilidad.
- Displasia espondiloepifisaria.
- Artrosis precoz.
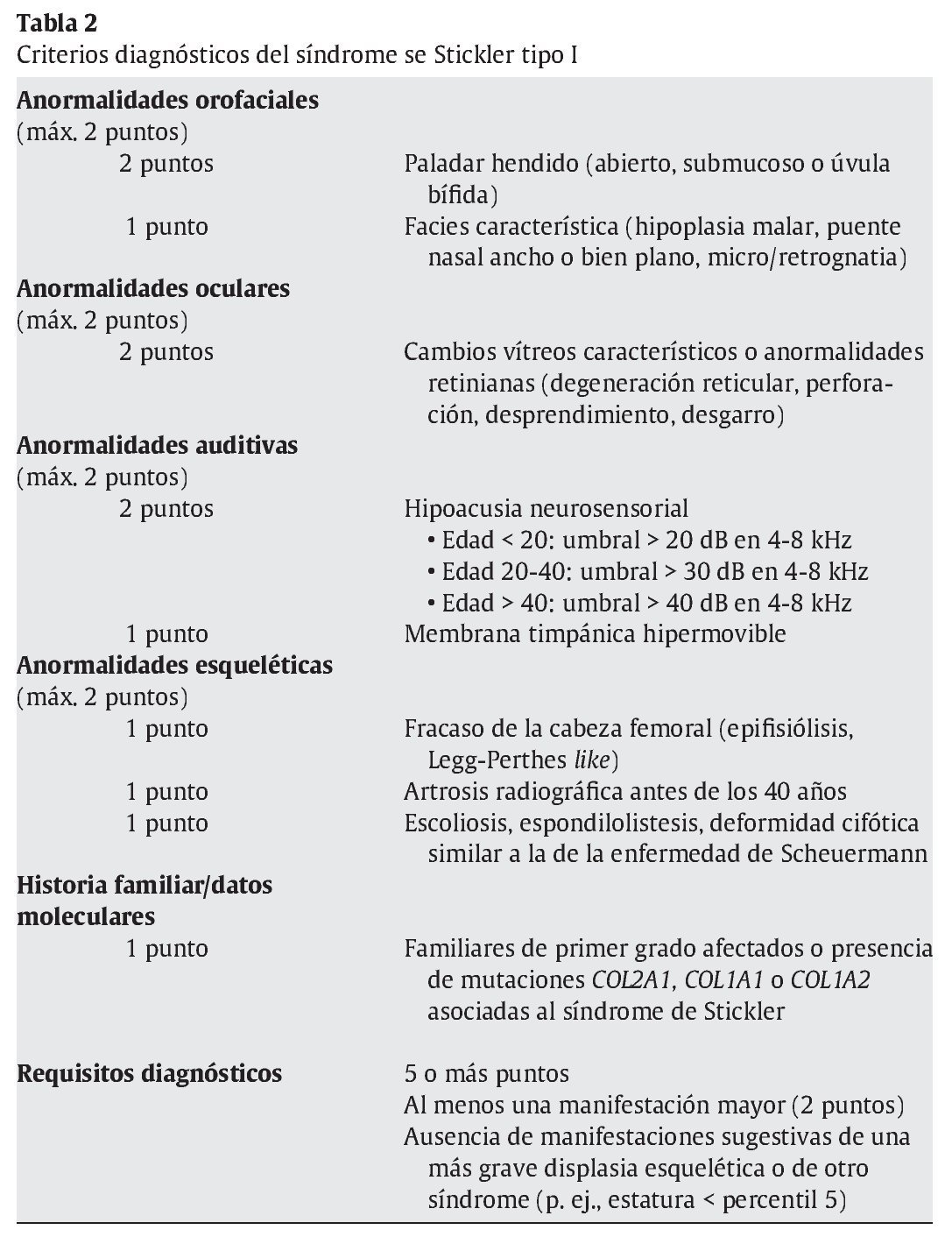
De los diferentes tipos de síndrome de Stickler descritos, el más frecuente es el tipo I (COL2A1), para el que Rose16 propuso criterios diagnósticos en el año 2005. Constan de criterios mayores, con un valor de 2 puntos, y criterios menores, con un valor de 1 punto. Se exige un total mínimo de 5 puntos (tabla 2).
Una vez establecida la sospecha clínica, se indaga en todos los miembros de la familia que sea posible. Si existen signos que nos puedan hacer sospechar la existencia de algún miembro afectado, debe ser sometido a un estudio básico, que comprende:
Valoración oftalmológica.
Valoración audiométrica.
Valoración cardiológica, que incluye un ecocardiograma al haberse descrito un incremento de la prevalencia de prolapso de la válvula mitral21, si bien no se ha confirmado en estudios posteriores22.
Valoración del aparato locomotor.
El diagnóstico diferencial se establece principalmente con otras enfermedades del colágeno con características clínicas similares a las del síndrome de Stickler15, entre las que destacan el síndrome de Wagner23, la miopía de alto grado15, el desprendimiento congénito de la retina24, la degeneración vitreorretiniana25, el síndrome de Binder26 o la secuencia de Robin12.
Una vez confirmada clínicamente la sospecha, es recomendable el estudio genético.
Tratamiento
La ausencia de un tratamiento etiológico hace que, actualmente, el único tratamiento posible sea el sintomático. En los primeros 12 meses de vida ya se actúa quirúrgicamente sobre los defectos del paladar duro cuando éstos existen. Oftalmológicamente, la corrección de los defectos de refracción es importante, así como la profilaxis del desprendimiento de retina27. Desde el punto de vista osteoarticular, se aconsejan las medidas habituales para disminuir el impacto de la artrosis precoz, y cuando deba recurrirse a la artroplastia, hay que tener muy en cuenta los problemas quirúrgicos derivados de la hipermovilidad articular existente en muchos casos. Se debe facilitar a los pacientes y a sus familiares toda la información y explicarles las posibles consecuencias de este trastorno genético. Es recomendable un control anual para detectar prematuramente las posibles complicaciones que puedan surgir.
* Autor para correspondencia.
Correo electrónico:grieramatute.girona.ics@gencat.cat (G. Riera Matute).
INFORMACIÓN DEL ARTÍCULO
Historia del artículo:
Recibido el 4 de febrero de 2009
Aceptado el 22 de abril de 2009