


Diversos estudios han demostrado que el colesterol libre (CL) hepático tiene una participación importante en la patogénesis de la esteatohepatitis no alcohólica (EHNA). Estos estudios han proporcionado evidencias de que la acumulación en el hígado de CL es tóxico a distintos niveles incluyendo: daño oxidativo a la mitocondria, estrés del retículo endoplasmático (RE) y activación de células de Kupffer (CKs) y células estelares hepáticas (CEH). En conjunto, estas evidencias sugieren que el contenido de CL hepático es importante en el inicio, mantenimiento y modulación de la respuesta inflamatoria asociada a la EHNA. En esta revisión, se discuten los distintos mecanismos participantes en la regulación de la homeostasis del colesterol y sus posibles implicaciones en el desarrollo y progresión del hígado graso no alcohólico (HGNA).
Several studies have shown that hepatic free cholesterol (FC) has an important role in the pathogenesis of nonalcoholic steatohepatitis (NASH). These studies have provided evidence that hepatic FC accumulation is toxic at different levels including: mitochondrial oxidative injury, endoplasmic reticulum (ER) stress, and activation of Kupffer cells (KCs) and hepatic stellate cell (HSCs). Altogether, this suggests that hepatic FC content is important for the initiation, maintenance and modulation of the inflammatory response associated with NASH. In this review several mechanisms that participate in the regulation of cholesterol homeostasis and their possible implications in the development and progression of nonalcoholic fatty liver disease (NAFLD) are discussed.
El hígado graso no alcohólico (HGNA) es la enfermedad hepática crónica más común, sin una historia de consumo significativo de alcohol. Esta patología está frecuentemente asociada con la presencia de obesidad, resistencia a la insulina y otras características del síndrome metabólico1,2. El HGNA se caracteriza por la acumulación de grasa en los hepatocitos que excede el 5% del peso del hígado3. El fenotipo histológico del HGNA se extiende desde grasa en el hígado (esteatosis simple, ES) hasta esteatohepatitis no alcohólica (EHNA)4. Esta última caracterizada por la presencia de esteatosis, inflamación lobular, balonamiento hepatocelular y diferentes grados de fibrosis, con un alto riesgo de desarrollar otras complicaciones como cirrosis y carcinoma hepatocelular3,4. Se ha establecido claramente que la obesidad central, la diabetes tipo 2 (DT2) y la resistencia a la insulina son factores de riesgo para el desarrollo de esteatosis hepática5. Sin embargo, los factores y mecanismos moleculares responsables para el desarrollo de inflamación en la progresión a EHNA, aún no han sido completamente dilucidados6,7. Se desconoce por qué algunos pacientes con HGNA permanecen con ES benigna, mientras que otros progresan a EHNA y fibrosis.
Prevalencia del HGNADe manera reciente, un estudio a nivel global encontró que el 25% de la población adulta en el mundo presenta HGNA8. Además, se estima que hasta un 30% de los sujetos con HGNA progresan a EHNA9. Es por ello que desde el año 2004, el número de adultos con complicaciones derivadas de EHNA, en espera de trasplante hepático (TH), casi se ha triplicado en los EUA, siendo actualmente la EHNA la segunda indicación más común para trasplante hepático, sólo después de la enfermedad hepática causada por el virus de la hepatitis C (VHC). Pero se estima que en los siguientes 10 a 20 años, las complicaciones por EHNA serán la primera causa de trasplante hepático10–12.
Patogénesis del HGNALa hipótesis más aceptada para entender la patogénesis del HGNA es la propuesta por Day y James (1998)13, que consiste en dos agresiones, la primera de ellas para el establecimiento de la esteatosis hepática, debido a la acumulación de ácidos grasos (AGs) y triglicéridos, como consecuencia del incremento en la captación y/o síntesis de AGs, así como la disminución de la beta oxidación de los mismos. La progresión de esteatosis a EHNA se establece a través de una segunda agresión, que parte de un hígado esteatótico más vulnerable al daño hepatocelular e inflamación. Los principales mecanismos involucrados en la segunda agresión incluyen al estrés oxidante, disfunción mitocondrial e inmunomodulación a través de distintas citocinas. Sin embargo, aun cuando esta hipótesis es ampliamente aceptada, estudios recientes sugieren que la progresión a EHNA ocurre en un proceso continuo, donde los lípidos con capacidad tóxica/pro-inflamatoria promueven un efecto de lipotoxicidad, lo cual conlleva a la progresión de esteatosis a EHNA14. A este respecto, distintas investigaciones se han enfocado en determinar la participación de diversos tipos de lípidos en la progresión a EHNA. Por ejemplo, aun cuando el HGNA se asocia con el incremento de triglicéridos hepáticos15, datos experimentales en modelos murinos con EHNA, en los cuales la síntesis de triglicéridos y la esteatosis hepática fue inhibida, el daño y la fibrosis hepática incrementó considerablemente16. Asimismo, pacientes con hipobetalipoproteinemia familiar (HBF) no progresaron a cirrosis a pesar de la presencia de esteatosis masiva17, lo que sugiere que la acumulación de triglicéridos per se podría no ser un factor determinante para la progresión. En este sentido, la investigación se ha enfocado en el estudio de otros lípidos como: ácidos grasos libres (AGL), diacilgliceroles, fosfolípidos (ceramidas y esfingolípidos) y más recientemente colesterol libre (CL)14. A este respecto, dos estudios a nivel lipidómico sugieren que son los niveles hepáticos de CL y no los triglicéridos, los que se encuentran diferencialmente elevados en los sujetos que progresan a EHNA18,19.
Función biológica del colesterolEl colesterol (3-hidroxi-5,6 colesteno) es un esterol (lípido) indispensable para la vida, desempeña funciones estructurales y metabólicas que son vitales para el ser humano20. Esta molécula se encuentra anclada estratégicamente en las membranas, donde modula la fluidez, permeabilidad y en consecuencia su función20. Constituye el elemento estructural básico del esqueleto de las membranas celulares21. Sin su refuerzo las membranas serían extremadamente fluidas y perderían su consistencia21. El colesterol también modula las funciones de las proteínas membranales y participa en varios procesos de tráfico y señalización transmembranal22. Además, es precursor de biomoléculas fisiológicamente importantes como las hormonas esteroideas, ácidos biliares y vitamina D20.
Síntesis, regulación y transporte del colesterolPara el aporte de colesterol corporal, la contribución de la síntesis de colesterol de novo en comparación con la ingesta dietética se estima en una proporción de ∼70:3022,23. En la práctica, esto probablemente varía considerablemente entre individuos, dependiendo tanto de la constitución genética (eficacia en la producción de colesterol en comparación con la absorción) y al suministro de colesterol en la dieta22.
Las dos principales fuentes del colesterol celular se derivan de la síntesis de novo y la captación de lipoproteínas plasmáticas. El colesterol se sintetiza a partir de acetil CoA a través de una serie de más de 30 reacciones enzimáticas24, las cuales pueden ser resumidas en tres etapas, la primera consiste en la síntesis de pirofosfato de isopentenilo, que es la unidad de construcción fundamental de colesterol; seguida de la condensación de seis moléculas de pirofosfato de isopentenilo para formar escualeno, y finalmente, el escualeno se cicla para formar lanosterol, que después de una serie de reacciones se convierte en colesterol24. La segunda fuente de colesterol celular ocurre a través de la captación de colesterol asociado a las lipoproteínas de baja densidad (LDL) mediante el receptor de las LDL (LDLR), pero también de colesterol en forma de lipoproteínas de alta densidad (HDL-C) y de remanentes de quilomicrones por receptores específicos22.
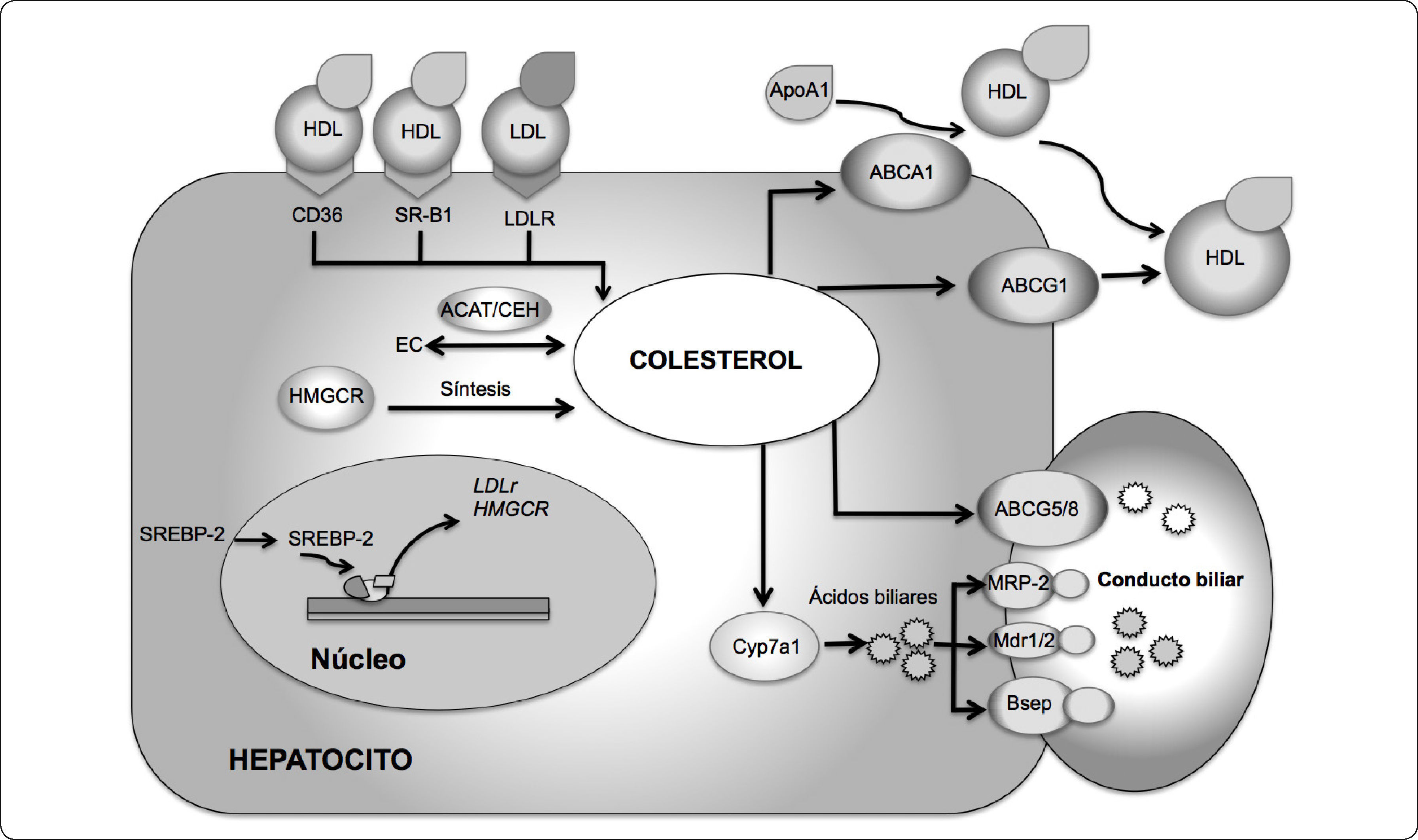
La homeostasis de colesterol intracelular, en los hepatocitos, se mantiene a través de una red coordinada que implica la participación de diversos genes involucrados en su captación, síntesis, bio-transformación, excreción y eflujo celular (Figura 1). La síntesis del colesterol está estrechamente regulada por el factor de transcripción de la proteína 2 de unión al elemento regulador de esteroles (SREBP-2). La forma madura de SREBP-2 inducida por SP-2 activa genes específicos en la biosíntesis del colesterol, tal es el caso de la enzima hidroximetilglutaril CoA reductasa (HMGCR), pieza clave en la regulación de la síntesis de colesterol22,25. De manera interesante, SREBP-2 también regula la captación hepática de colesterol, a través de la expresión hepática del receptor LDLR25,26. De tal manera, que cuando las células requieren colesterol, el factor de transcripción SREBP-2 aumenta la expresión de ambos, la HMGCR y el LDLR, resultando en un incremento en la síntesis y captación de colesterol27 (Figura 1).
, síntesis (HMGCR), bio-transformación (CYP7A1), eflujo celular al conducto biliar (ABCG5/G8, MRP-2, Mdr1/2 y Bsep), eflujo celular hacia las HDL (ABCA1/G1); así como la regulación por factores de transcripción que censan los niveles de colesterol intracelular (SREBP-2). CD36, Proteína de transporte de ácidos grasos; SR-B1, Receptor basurero clase B tipo 1; LDL-R, Receptor de las lipoproteínas de baja densidad; HMGCR, Hidroximetilglutaril CoA reductasa; Cyp7a1, Colesterol 7 alfa-hidroxilasa; ABCG5, Transportador G5 dependiente de unión a ATP; ABCG8, Transportador G8 dependiente de unión a ATP; MRP-2, Proteína asociada a resistencia a multidrogas-2; Mdr1/2, Proteína de resistencia a multidrogas-1/2, Bsep, Proteína 2 exportadora de sales biliares; ABCA1, Transportador A1 dependiente de unión a ATP, ABCG1, Transportador G1 dependiente de unión a ATP; SREBP-2, Proteína 2 de unión al elemento regulador de esteroles; ACAT2, la acetil- CoA acetiltransferasa 2; CEH, la colesterol éster hidrolasa; EC, ésteres de colesterol. Basado en 28,29.")
Homeostasis del colesterol. La homeostasis de colesterol intracelular está coordinada por diferentes vías metabólicas, por ejemplo: captación (CD36, SR-B1, LDL-R), síntesis (HMGCR), bio-transformación (CYP7A1), eflujo celular al conducto biliar (ABCG5/G8, MRP-2, Mdr1/2 y Bsep), eflujo celular hacia las HDL (ABCA1/G1); así como la regulación por factores de transcripción que censan los niveles de colesterol intracelular (SREBP-2). CD36, Proteína de transporte de ácidos grasos; SR-B1, Receptor basurero clase B tipo 1; LDL-R, Receptor de las lipoproteínas de baja densidad; HMGCR, Hidroximetilglutaril CoA reductasa; Cyp7a1, Colesterol 7 alfa-hidroxilasa; ABCG5, Transportador G5 dependiente de unión a ATP; ABCG8, Transportador G8 dependiente de unión a ATP; MRP-2, Proteína asociada a resistencia a multidrogas-2; Mdr1/2, Proteína de resistencia a multidrogas-1/2, Bsep, Proteína 2 exportadora de sales biliares; ABCA1, Transportador A1 dependiente de unión a ATP, ABCG1, Transportador G1 dependiente de unión a ATP; SREBP-2, Proteína 2 de unión al elemento regulador de esteroles; ACAT2, la acetil- CoA acetiltransferasa 2; CEH, la colesterol éster hidrolasa; EC, ésteres de colesterol. Basado en 28,29.
Por otra parte, el procesamiento del colesterol tiene lugar en diferentes localizaciones subcelulares; por lo tanto, la entrega de colesterol entre estos sitios es un medio importante para regular estas reacciones22. El colesterol se transfiere entre las membranas subcelulares por transporte vesicular y por mecanismos no vesiculares22. Las vesículas transportan componentes de la membrana a los orgánulos sub-celulares por medio de pistas en el citoesqueleto, mientras que los mecanismos de transferencia de colesterol no-vesiculares presumiblemente utilizan proteínas de transferencia de lípidos en el citosol22.
En este sentido, la homeostasis del colesterol debe ser finamente regulada para mantener los niveles de colesterol en rangos adecuados y realice correctamente sus funciones biológicas.
El alto consumo de colesterol en la dieta aumenta el riesgo de desarrollar HGNAEl exceso en el consumo de colesterol se ha considerado como una causa fundamental del HGNA30–32. En modelos animales se ha observado que una dieta alta en colesterol, dentro de un rango de energía normal, induce el comienzo del HGNA y su progresión a otras complicaciones como inflamación, EHNA y fibrosis33–35. Por otra parte, en un estudio que incluyó un número amplio de pacientes, el consumo de colesterol en la dieta se asoció independientemente a mayor riesgo de cirrosis y cáncer hepático, mientras que el consumo de grasa total no mostró diferencias36. Además, una encuesta nutricional encontró que la ingesta de colesterol en la dieta fue significativamente mayor en pacientes con EHNA, que en sujetos saludables37. Otro grupo de investigadores encontró en pacientes obesos y no obesos, con HGNA, que la ingesta del colesterol fue significativamente más alta en los pacientes con HGNA, que en sujetos saludables38. En este mismo estudio, llama la atención que los pacientes no obesos con HGNA ingirieron más colesterol que los pacientes con HGNA obesos38, lo cual indica que la ingesta de colesterol en la dieta es esencial para el inicio y progresión del HGNA independiente de la obesidad39.
Factores genéticos en el metabolismo del colesterol se asocian a HGNADiversos factores genéticos que afectan el metabolismo del colesterol se han asociado a la progresión de esteatosis y EHNA. De manera muy interesante, en un estudio de seguimiento a 7 años, en sujetos sin presencia de obesidad o diabetes, se observó que los sujetos con el polimorfismo rs133291 (C/T) en el gen SREBP-2 predice la incidencia de HGNA, y la predisposición para desarrollar EHNA40. Además, de manera reciente se reportó la asociación del polimorfismo rs2228314 G>C de este mismo gen, con el desarrollo de HGNA41. Por tanto, polimorfismos que afecten la función adecuada de SREBP-2 podrían exacerbar el efecto tóxico del colesterol, aun en ausencia de un consumo excesivo de este lípido en la dieta.
De manera similar, se ha observado que un polimorfismo en la farnesil- difosfato farnesil transferasa 1 (FDFT1), que codifica para la escualeno sintasa –enzima que cataliza el primer paso en la biosíntesis de esteroles– se ha asociado a EHNA42. A su vez, la deficiencia de la lipasa ácida lisosomal que es causada por mutaciones en el gen LIPA, desencadena esteatosis, fibrosis y cirrosis debido a la acumulación de esteres de colesterol y triglicéridos en los hepatocitos y en células de Kupffer (CKs)43,44. Recientemente, un estudio demostró que el reemplazamiento de este defecto, con una proteína LIPA recombinante, revierte este efecto en humanos45.
Lo anterior sugiere que el componente genético de esta patología en combinación con los factores ambientales, por ejemplo el alto consumo de colesterol en la dieta, podrían explicar, al menos en parte, la susceptibilidad para desarrollar HGNA y EHNA.
DESEQUILIBRIO EN LA HOMEOSTASIS DEL COLESTEROL HEPÁTICO EN EL HGNALos estudios en modelos animales con obesidad, diabetes o dislipidemias demuestran que el desequilibrio en la homeostasis de colesterol promueve su acumulación y modula la transición de esteatosis a EHNA, lo que correlaciona directamente con el grado de daño hepático, muerte celular por apoptosis, acumulación de macrófagos y fibrosis28. Además, Min y colaboradores (2012) encontraron en pacientes con EHNA que distintas rutas metabólicas involucradas en la homeostasis del colesterol hepático se encuentran desreguladas29.
Biosíntesis de colesterolLa HMGCR es la enzima limitante en la velocidad de la síntesis de colesterol46. Distintos estudios han revelado que la expresión y actividad de la HMGCR se incrementa en el hígado de pacientes con EHNA, y parece estar asociado a una disminución en los niveles de fosforilación de su forma activa18,29. También, se ha demostrado que la expresión de la HMGCR correlaciona directamente con la acumulación del colesterol y el daño hepático en sujetos con HGNA29, y revela a la HMGCR como posible pieza clave en el desarrollo a EHNA, a través de la acumulación de colesterol.
Captación de lipoproteínas ricas en colesterolAdemás del receptor de las lipoproteínas de baja densidad (LDLR), el colesterol también puede ser captado a través del receptor basurero clase B tipo 1 (SR-B1) y la proteína de transporte de ácidos grasos CD36, las cuales funcionan como receptores de las lipoproteínas de alta densidad (HDL)47,48 (Figura 1). Diversos estudios demuestran que el receptor LDLR se encuentra sobre-regulado en modelos experimentales con esteatosis y EHNA, que podría contribuir al incremento en el contenido del colesterol hepático28,49; sin embargo, recientemente un estudio encontró que la expresión hepática del receptor LDLR decreció en sujetos con EHNA, aunado a un incremento en los niveles plasmáticos de las LDLs29. Por tanto, se requieren otros estudios para definir si el receptor LDLR participa en el desarrollo del HGNA. A su vez, la expresión hepática del receptor CD36 se encuentra sobre-regulada y correlaciona con la severidad de la esteatosis en pacientes con EHNA50. Estos hallazgos son congruentes con lo reportado en otro estudio experimental, en donde la ausencia de CD36, protege del desarrollo de esteatosis e inflamación hepática51. Por otra parte, la expresión del receptor SR-B1 se encuentra incrementada en modelos experimentales con HGNA52, lo cual sugiere un incremento en la captación de colesterol; sin embargo, otro estudio muestra que la transcripción de SR-B1 se encuentra disminuida en EHNA en el modelo murino28. Estudios futuros serán necesarios para clarificar esta discrepancia.
Regulación de colesterol por SREBP-2SREBP-2 es un factor de transcripción clave en la regulación de los niveles de colesterol en estados de resistencia a la insulina e hiperinsulinemia, ambos comunes en los pacientes con esteatosis y EHNA. Llama la atención que la expresión hepática de SREBP-2 está incrementada en pacientes con EHNA18, y es consistente con otros casos reportados28,29,53. Recientemente, un estudio demostró también que SREBP-2 contribuye a la acumulación de CL en células estelares hepáticas (CEHs), lo que condujo al desarrollo de EHNA y fibrosis53. Por tanto, estos resultados sugieren que SREBP-2 juega un papel importante en la progresión a EHNA y fibrosis, por lo que futuras investigaciones serán requeridas para determinar si SREBP-2 podría ser un blanco terapéutico para la prevención y/o tratamiento de la EHNA y fibrosis.
PPARs en la homeostasis del colesterolDiversos estudios indican que la homeostasis del colesterol puede ser modulada por el efecto de los receptores activados por proliferadores de peroxisomas (PPARs)54–56. De hecho, se ha observado en modelos murinos con procesos inflamatorios hepáticos, que los niveles de expresión de PPAR-α se encuentran reducidos, sugiriendo una disminución en el eflujo de colesterol, a través de la regulación de la expresión de su gen blanco, el transportador de colesterol A1 dependiente de unión a ATP (ABCA1, por sus siglas en inglés)57. Además, recientemente en un modelo de ratón carente de PPAR-α, los niveles de colesterol aumentaron en el hígado, aunado a un incremento en el daño hepático58. De manera adicional, en el humano, se ha observado que los niveles de expresión hepática de PPAR-α correlacionan inversamente con la severidad de la EHNA59. Lo anterior sugiere que PPAR-α, en parte, a través de la regulación de la homeostasis del colesterol, podría ser un blanco terapéutico potencial en el tratamiento de la EHNA.
Por otra parte, se ha observado también en CEHs activadas, que la supresión de PPAR-γ disminuye la expresión del gen 1 inducido por insulina (Insig-1, por sus siglas en inglés), cuyo resultado es la disrupción del sistema de retro-alimentación en la homeostasis del colesterol, mediado por SREBP-253. Lo anterior conduce al incremento del colesterol libre en las CEHs, y su posterior activación; lo que desencadena procesos fibrogénicos y EHNA53.
Transporte de colesterol intracelularEl colesterol es insoluble en agua, por tanto, requiere ciertos mecanismos especializados para moverse a través de los organelos en el citosol. Se ha sugerido que el desequilibrio en estos mecanismos aumenta la acumulación de CL y la toxicidad en ciertos compartimentos celulares, lo que promueve el desarrollo de la EHNA60,61.
La caveolina-1 es la principal proteína estructural de las caveolas, que son bolsas lipídicas con alto contenido de colesterol y esfingolípidos62. Además, la caveolina-1 juega una participación importante en el transporte intracelular del colesterol62. Consistente con ello, un modelo experimental con HGNA mostró que los niveles de expresión de la caveolina-1 aumentaron significativamente alrededor y dentro de las gotas de lípidos (GLs), así como dentro de la membrana interna de la mitocondria63. Estos hallazgos sugieren que la caveolina-1 participa en la lipogénesis anormal y en la función mitocondrial típica de los hepatocitos esteatóticos en el HGNA.
Las proteínas Niemannn-Pick C1 y C2 (NPC1 y NPC2) se encuentran en las membranas de los lisosomas y los endosomas tardíos; presentan una participación crítica en la regulación del tráfico de colesterol intracelular, de los compartimentos endolisosomales al resto de la célula64. Mutaciones en los genes Ncp1 y Ncp2 se relacionan con la enfermedad Niemannn-Pick65–67, que es una enfermedad caracterizada por la acumulación de CL en la mayoría de los tejidos incluido el hígado65,67. De manera interesante, se ha observado que la deficiencia de NPC1/2 conduce a HGNA, ganancia de peso y síndrome metabólico en modelos experimentales68. Lo que permite sugerir que NCP1 y 2 podrían contribuir al desarrollo del HGNA.
La familia de proteínas de transferencia de lípidos relacionadas a StAR (START) está implicada en la transferencia de lípidos intracelulares69. Particularmente, la proteína de regulación aguda de esteroidogénesis (StAR) facilita el movimiento de colesterol de los almacenamientos celulares a la mitocondria70. De manera interesante, Caballero y colaboradores (2009) encontraron que los niveles de mRNA de StAR están incrementados 7 y 15 veces en pacientes con esteatosis y EHNA18. Lo que sugiere que la acumulación de CL en la mitocondria, regulado por StAR, podría jugar un papel importante en la progresión de la enfermedad. De manera similar, la proteína endosomal MLN64 (por sus siglas en inglés, metastatic lymph node protein 64) modula el eflujo de colesterol de los endosomas a la mitocondria71. Por lo que a MLRG4 se le ha propuesto como una proteína candidato, en el aumento del transporte del colesterol a la mitocondria, y en el desarrollo del HGNA60. De hecho, la sobre-expresión de MLRG4 induce un incremento en los niveles de CL hepático, asociada con apoptosis y daño en el hígado72. Sin embargo, en otro estudio se observó que los niveles de expresión hepática de MLRG4 disminuyeron en ratones obesos modificados genéticamente, y los autores proponen que MLRG4 podría proteger al hígado del efecto lipotóxico.
Otra proteína que parece ser importante en la regulación del tráfico de colesterol intracelular, es la proteína de unión a oxiesteroles (OSBP, por sus siglas en inglés) y proteínas asociadas a OSBP. OSBP es una proteína citosólica que se le ha implicado en la regulación del colesterol celular, esfingomielina, y transporte de oxiesteroles73. Aunque no hay una conexión directa entre los miembros de la familia OSBP y el HGNA, un estudio reveló que ORP8 –un miembro de esta familia– juega un papel importante en la homeostasis de lípidos74 y también la sobre-expresión de ORP8 redujo los niveles de colesterol y triglicéridos hepáticos, mientras que el silenciamiento los incrementó a través de la modulación de la expresión de la proteína 1 y 2 de unión al elemento regulador de esteroles (SREBP-1 y 2)74. Por tanto, investigaciones adicionales serán requeridas para definir la participación de las proteínas OSBP en el desarrollo del HGNA.
Eflujo de colesterol y transportadores de colesterol ABCA1/G1El transportador de colesterol ABCA1, es una proteína integral de la membrana, que participa en el transporte reverso del colesterol. Esta proteína se ha estudiado ampliamente, debido a su estrecha relación con las concentraciones plasmáticas del HDL-C. La lipidación de apoA-1 mediante el transportador ABCA1 es un paso limitante en la velocidad del transporte reverso del colesterol y la generación de HDLs en el plasma75. Por ello, un transporte eficiente o eflujo de colesterol podría ser clave en la prevención del desarrollo a EHNA. Recientemente, se reportó que la sobre-expresión de la proteína de ABCA1 disminuye significativamente el contenido de ácidos grasos libres, triglicéridos y colesterol en células hepáticas, mientras que el silenciamiento de ABCA1 incrementa los niveles de estos lípidos76. Además, se sabe que el hígado de ratas con EHNA expresan niveles bajos de la proteína ABCA1, pero no de los niveles de expresión del mRNA77. De manera similar, se encontró que sólo los niveles de la proteína ABCA1 se encuentran reducidos en sujetos con EHNA78. Lo cual sugiere mecanismos de regulación post-transcripcional regulando los niveles de la proteína ABCA1 en la EHNA78. En este estudio, el nivel de expresión de miR-33a –un regulador post-trancripcional de ABCA1– correlacionó negativamente con los niveles de la proteína ABCA1 y positivamente con la severidad de la enfermedad78. Lo que podría explicar la discrepancia observada entre los niveles de mRNA y proteína. Con base en los resultados anteriores, se sugiere que ABCA1 podría contribuir a la patogénesis del HGNA, posiblemente a través de la modulación del colesterol.
Por otra parte, se ha observado en modelos murinos, que la ausencia del transportador de colesterol G1 dependiente de unión a ATP (ABCG1, por sus siglas en inglés) promueve la acumulación de lípidos en hepatocitos y macrófagos79. Además, ABCG1 promueve el eflujo de ciertos oxi-esteroles lipotóxicos (como el 7-cetocolesterol)80. De manera interesante, se ha encontrado que los niveles de la proteína ABCG1 se encuentran reducidos en sujetos con EHNA45,78. Por tanto, ABCG1 podría contribuir al HGNA a través de la modulación del colesterol. Sin embargo, se requieren otros estudios, en modelos experimentales, que definan si la disminución de ABCG1 es causa de EHNA.
Excreción y bio-transformación de colesterolLa regulación de los niveles de colesterol también implica su excreción al conducto biliar a través de diferentes vías, en las que participan los transportadores de colesterol G5 y G8 dependientes de unión a ATP (ABCG5 y ABCG8, por sus siglas en inglés)75,81 (Figura 1). De manera interesante, ratones que carecen de los transportadores ABCG5/8 muestran una reducción en la secreción de colesterol al conducto biliar y son más susceptibles al desarrollo de esteatosis, resistencia a la insulina hepática, y pérdida del control de la glucemia cuando son alimentados con un dieta alta en grasa82. Además, Min y colaboradores (2012) encontraron que los niveles de expresión de ABCG8 se encuentran reducidos en sujetos con EHNA29. Estos resultados soportan la hipótesis de que la reducción en el eflujo del colesterol a través del conducto biliar, vía ABCG5/8, podría contribuir al desarrollo de la EHNA.
De manera similar, la bio-transformación hepática del colesterol en ácidos biliares (ABs) constituye la principal ruta del catabolismo del colesterol, en donde la colesterol 7-alfa-hidroxilasa (Cyp7a1) es la enzima limitante en la velocidad de producción del 90% de los ABs a partir de colesterol (Figura 1). De manera interesante, niveles reducidos de Cyp7a1 se observaron en un modelo de rata con EHNA inducido por incremento de colesterol en la dieta83. Además, los niveles de proteína de Cyp7a1 y Cyp27a se encuentran reducidos en sujetos con EHNA29. Por tanto, la reducción en la bio-transformación del colesterol podría también contribuir a la acumulación del colesterol hepático, y posiblemente a la progresión a EHNA.
Esterificación y des-esterificación de colesterolLa relación dinámica entre ésteres de colesterol (EC) y CL está dada por la esterificación y des-esterificación llevada acabo por dos enzimas del RE: la acetil- CoA acetiltransferasa 2 (ACAT2) y la colesterol éster hidrolasa (CEH). Aunque la participación de ACAT2, en el desarrollo del HGNA, parece no ser concluyente, debido a la discrepancia de los resultados encontrados18,28,29,84, los niveles de expresión de CEH se encuentran incrementados en un estudio que incluye pacientes con EHNA29. Estos hallazgos sugieren que CEH contribuye al incremento en el contenido de CL, mediante la inducción de la hidrólisis de EC, aumentando el contenido de CL, lo que podría desencadenar EHNA.
MicroRNAs en la modulación del colesterol hepáticoRecientemente, además de la regulación transcripcional clásica, una clase de RNAs no codificantes denominados microRNAs (miRNAs), constituidos de ∼23 nt de RNA endógeno, se han presentado como reguladores de la expresión y actúan predominantemente a nivel post-transcripcional85,86. En la última década, se ha demostrado de manera más clara y progresiva que los microRNAs funcionan como reguladores importantes de un amplio rango de procesos celulares, incluyendo el metabolismo del colesterol 85,87,88 y se ha identificado que la alteración en los perfiles de los microRNAs está asociada con esteatosis y EHNA89–91.
De manera interesante, distintos microRNAs se han relacionado a EHNA y al metabolismo del colesterol hepático, lo que sugiere que estas micro-moléculas podrían contribuir a la progresión de la EHNA a través de la modulación de los niveles de colesterol hepático. Por ejemplo, miR-34a se le ha implicado como un factor central en el desarrollo de la EHNA91. El mejor blanco caracterizado para miR-34a es la sirtruina 1 (SIRT1), una desacetilasa dependiente de NAD, la cual disminuye los niveles de la HMGCR fosforilada29. De manera interesante, miR-34a se encuentra sobre-expresado en sujetos con EHNA89,92. Además, se ha sugerido que el incremento de miR-34a puede modular la fosforilación de la HMGCR y mantenerla en su forma activa, aumentando los niveles de CL hepático29. Por tanto, estos resultados sugieren que cambios en la expresión de miR-34a podrían contribuir al desarrollo de la EHNA, al menos en parte, a través de la modulación de la HMGCR.
Otro microRNA de relevancia es miR-122, el microRNA más abundante en el hígado y comprende aproximadamente el 70% de los microRNAs expresados en él93,94. Encontrándose que miR-122 es un regulador clave en el metabolismo del colesterol y los ácidos grasos95,96. Llama la atención que miR-122 se encuentra disminuido en sujetos con EHNA89,92. De manera interesante, estudios funcionales demostraron que el silenciamiento de miR-122, en células HepG2, conduce a un incremento inicial de sus genes blanco: Hmgcr y Srebp-2, que son claves en la síntesis del colesterol89. De manera contradictoria, en modelos murinos, se ha observado que el silenciamiento de miR-122 resulta en una reducción del 25-30% en los niveles de colesterol plasmático, aunado a una reducción hepática en la expresión de genes que participan en la síntesis del colesterol: 3-hidroxi-metilglutaril-CoA sintetasa 1 (Hmgcs1), 7- dehidrocolesterol reductasa (Dhcr7) y Hmgcr97. En un modelo de ratón alimentado con una dieta alta en grasa, se ha observado que la inhibición de miR-122 induce una disminución significativa en la síntesis de colesterol y ácidos grasos95. Por tanto, lo anterior sugiere que la regulación de miR-122 en humanos es distinta al modelo murino, y será de relevancia determinar en primates no humanos el efecto de la inhibición o inducción de miR-122 en el desarrollo del HGNA.
Recientemente, se ha demostrado que miR-21 se encuentra disminuido en el modelo de ratón y en sujetos con HGNA98,99. Además, en modelos in vitro con HGNA, se ha observado que la disminución de miR-21 aumenta la expresión de su gen blanco la Hmgcr; por el contrario, análogos de miR-21 disminuyen los niveles de TG, CL y colesterol total99. Por tanto, estos resultados sugieren que miR-21 contribuye a la modulación del colesterol hepático, y posiblemente al desarrollo del HGNA.
Mir-33 es otro microRNA que está involucrado en la regulación de la homeostasis del colesterol y oxidación de los ácidos grasos100,101. En humano se han identificado dos isoformas de miR-33: miR-33a y miR-33b; mientras que en ratón, hay sólo un gen miR-33, conservado con el miR-33a de humano100,101. El gen miR-33a está localizado en el intrón 16 del gen Srebp-2 y miR-33b está presente en el intrón 17 del gen Srebp-1100,101. En una gran cantidad de estudios se ha demostrado que la sobre-expresión de miR-33, en hepatocitos y macrófagos, resulta en la disminución de la expresión de ABCA1 y del eflujo de colesterol101–104. De igual manera, la inhibición endógena de miR-33, en el mismo tipo de células, promueve un incremento tanto en la expresión de ABCA1 como en el eflujo de colesterol101–103. Estos resultados son congruentes en estudios de primates no humanos, en donde la inhibición de miR-33 aumentó los niveles de expresión de ABCA1 y el eflujo de colesterol105. Recientemente, se observó que los niveles de expresión hepática de miR-33a, pero no miR-33b, correlacionaron negativamente con la expresión de su gen blanco Abca1 en biopsias de pacientes con HGNA, además, miR-33a aumentó de manera progresiva con el desarrollo a EHNA78. Este resultado es consistente con lo publicado por Lendvai y colaboradores (2014) en donde los niveles de miR-33a se encontraron elevados en sujetos con HGNA106. Además, estudios recientes, en modelos murinos, sugieren que la acumulación de CL en CEHs promueve el desarrollo de EHNA a fibrosis hepática, posiblemente a través del incremento de los niveles de miR-33a, lo que podría conllevar a la supresión de los niveles de ABCA1 y del eflujo de colesterol53. Con base en lo anterior, miR-33a podría contribuir a la acumulación del colesterol hepático, al desarrollo de la EHNA y fibrosis, al menos en parte, a través de la regulación de ABCA1. Por tanto, será de relevancia definir si miR-33a está involucrado en la progresión de EHNA a fibrosis hepática en el humano, y si una modulación terapéutica de miR-33a podría ser útil para el tratamiento de la fibrosis.
El microRNA-144 también regula la expresión hepática de ABCA1 en modelos murinos, y en cultivos celulares de macrófagos humanos, promueve la acumulación de colesterol hepático, atenuando el eflujo de colesterol a través de apo-A1107–109 siendo consistente con la asociación de miR-144 con los niveles de la proteína ABCA1 y con niveles incrementados en sujetos con EHNA78.
Lo anterior sugiere que un desequilibrio en la vías metabólicas, que participan en la homeostasis del colesterol en el hígado, podría promover la acumulación de colesterol, y desencadenar daño hepático a distintos niveles celulares, promoviendo la progresión de EHNA y fibrosis.
EL COLESTEROL EN LA TOXICIDAD CELULAR Y PATOGÉNESIS DEL HGNAToxicidad del colesterol en el HGNAA diferencia de la membrana plasmática (MP) que contiene altas concentraciones de colesterol, el retículo endoplasmático (RE) y la mitocondria poseen una cantidad menor de colesterol (3-5% del colesterol celular total)110 y son altamente sensibles a la pérdida de fluidez membranal ocasionada por enriquecimiento de colesterol111.
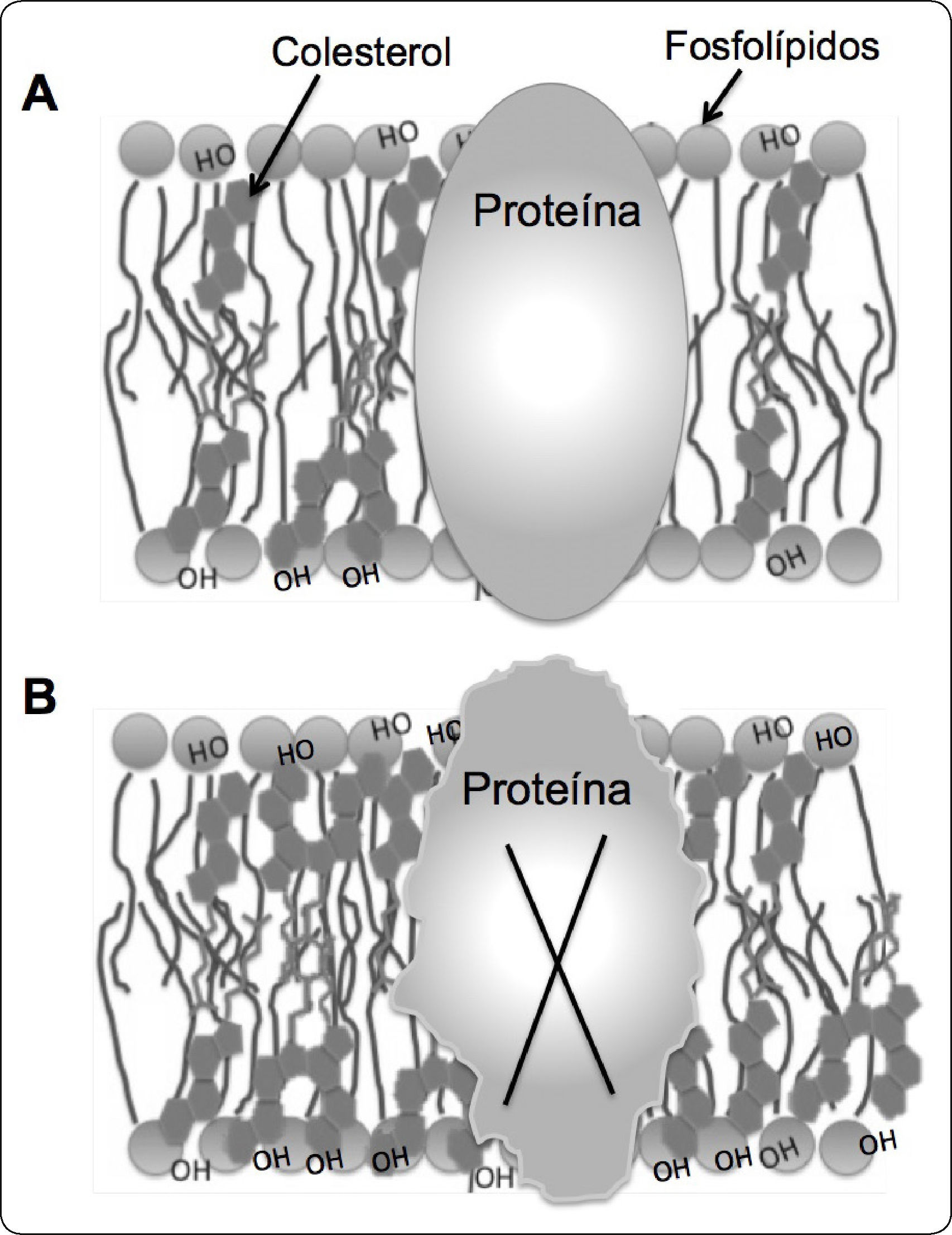
Se ha demostrado que niveles elevados de CL afectan la organización y las propiedades físicas de las membranas mitocondriales y del RE, lo que disminuye la fluidez de las membranas112. Estos eventos afectan negativamente a ciertas proteínas transmembranales, que requieren libertad conformacional para realizar sus funciones de manera apropiada113 (Figura 2).
 El colesterol se encuentra anclado estratégicamente en las membranas de la célula, donde modula la fluidez y permeabilidad. La molécula de colesterol constituye el elemento estructural básico del esqueleto de las membranas celulares, sin su refuerzo las membranas serían extremadamente fluidas y perderían su consistencia. (B) El aumento de los niveles de colesterol en la membrana afecta la organización y sus propiedades físicas, lo que disminuye la fluidez y aumenta la rigidez. En consecuencia, estos eventos afectan negativamente a ciertas proteínas transmembranales, las cuales requieren libertad conformacional para realizar sus funciones de manera apropiada. Basado en 21,112.")
El aumento de colesterol en la membrana altera su estructura. (A) El colesterol se encuentra anclado estratégicamente en las membranas de la célula, donde modula la fluidez y permeabilidad. La molécula de colesterol constituye el elemento estructural básico del esqueleto de las membranas celulares, sin su refuerzo las membranas serían extremadamente fluidas y perderían su consistencia. (B) El aumento de los niveles de colesterol en la membrana afecta la organización y sus propiedades físicas, lo que disminuye la fluidez y aumenta la rigidez. En consecuencia, estos eventos afectan negativamente a ciertas proteínas transmembranales, las cuales requieren libertad conformacional para realizar sus funciones de manera apropiada. Basado en 21,112.
Estudios realizados, en modelos murinos y en cultivos primarios de hepatocitos, demostraron que el exceso de CL en la mitocondria hace sensible al hepatocito a citocinas a través de la depleción del glutatión mitocondrial (mGSH, por sus siglas en inglés), que es esencial para controlar la generación de especies reactivas de oxígeno (ERO). Ésto sucede a través del incremento del CL en la mitocondria que reduce la fluidez de su membrana y afecta la estabilidad de la proteína transmembranal acarreadora de 2-oxoglutarato, que transporta glutatión del citosol al interior de la mitocondria114. En consecuencia, los niveles de glutatión reducen en la mitocondria, y los hepatocitos se vuelven sensibles a citocinas inflamatorias como el factor de necrosis tumoral α (TNFα, por sus siglas en inglés) y la molécula pro-apoptótica Fas. Lo anterior promueve la permeabilización de la membrana mitocondrial, la liberación del citocromo C, necrosis y apoptosis; e inflamación hepática y EHNA en modelos experimentales114.
Estrés en el retículo endoplasmático (RE) inducido por colesterolAlgunos estudios sugieren que la acumulación del colesterol en la membrana del RE altera sus funciones y su capacidad para el correcto plegamiento de proteínas115,116, ésto resulta en una condición conocida como estrés del RE o respuesta a proteínas mal plegadas (UPR, por sus siglas en inglés). Se sabe que esta condición juega un papel importante en desórdenes asociados a la obesidad, incluyendo el HGNA117. Un ejemplo de ello, son los desórdenes conformacionales de la ATPasa de Ca2+ del retículo sarco(endo)plásmico (SERCA, por sus siglas en inglés, ER sarco(endo) plasmic reticulum Ca2+-ATPase), que es altamente sensible a incrementos mínimos en los niveles de colesterol en el RE, y afecta su actividad ATPasa118,119. En consecuencia las altas concentraciones fisiológicas de Ca2+ dentro del RE disminuyen y como resultado, se pierde la capacidad del correcto plegamiento de proteínas; lo que conduce a estrés en RE118,119. Sin embargo, aunque se ha descrito la importancia de la acumulación del CL en el RE en hepatocitos115,116, aún se desconoce la implicación que pudiera tener en el desarrollo a EHNA.
Inducción de células de Kupffer (CKs) y activación de células estelares hepáticas (CEHs) por colesterolLas células de Kupffer (CKs) representan un 20-25% de las células no-parenquimatosas del hígado. Además de su participación, bien conocida como células fagocíticas, las CKs activadas representan una fuente principal de citocinas pro-inflamatorias y pro-fibrogénicas (TNF-α y el factor de crecimiento transformante beta 1, TGF-β1)61. Asimismo, se ha demostrado, en modelos murinos con una dieta alta en grasa y colesterol, que la acumulación de CL en CKs es requerida para la activación de un fenotipo pro-inflamatorio, lo cual conduce al desarrollo de EHNA120. Por otra parte, debido a que las CKs no sintetizan colesterol, ellas lo obtienen principalmente a través de la captación de las lipoproteínas de baja densidad en estado oxidado (oxLDLs). Llama la atención, que la acumulación de colesterol lisosomal en las CKs promueve una respuesta inflamatoria, y causa incremento en la inflamación hepática en modelos experimentales con EHNA121,122. Se ha observado que el efecto de ciertos agentes, que reducen la acumulación de colesterol en los lisosomas, da como resultado una reducción en la inflamación hepática en modelos experimentales con EHNA123,124. Sin embargo, se requerirán más estudios para definir si la captación sobre-regulada de las oxLDL en CKs conduce a la acumulación de colesterol, y promueve un respuesta inflamatoria en el desarrollo del HGNA en el humano.
Por otra parte, la activación de las células estelares hepáticas (CEHs) participa en procesos fibrogénicos en el desarrollo del HGNA125. Evidencias recientes en modelos murinos, revelaron que la acumulación de CL intracelular activa directamente las CEHs, y las vuelve sensibles a la inducción por TGF-β. En consecuencia, se disparan procesos fibrogénicos mediados por el receptor tipo toll-4 (TLR4, por sus siglas en inglés), lo que conduce al incremento de la fibrosis en la EHNA53,126. Recientes investigaciones muestran que la eliminación de la acetil-CoA acetiltransferasa 1 (ACAT1), principalmente expresada en CEHs, exacerba la fibrosis hepática a través de la acumulación de CL en este tipo de células127.
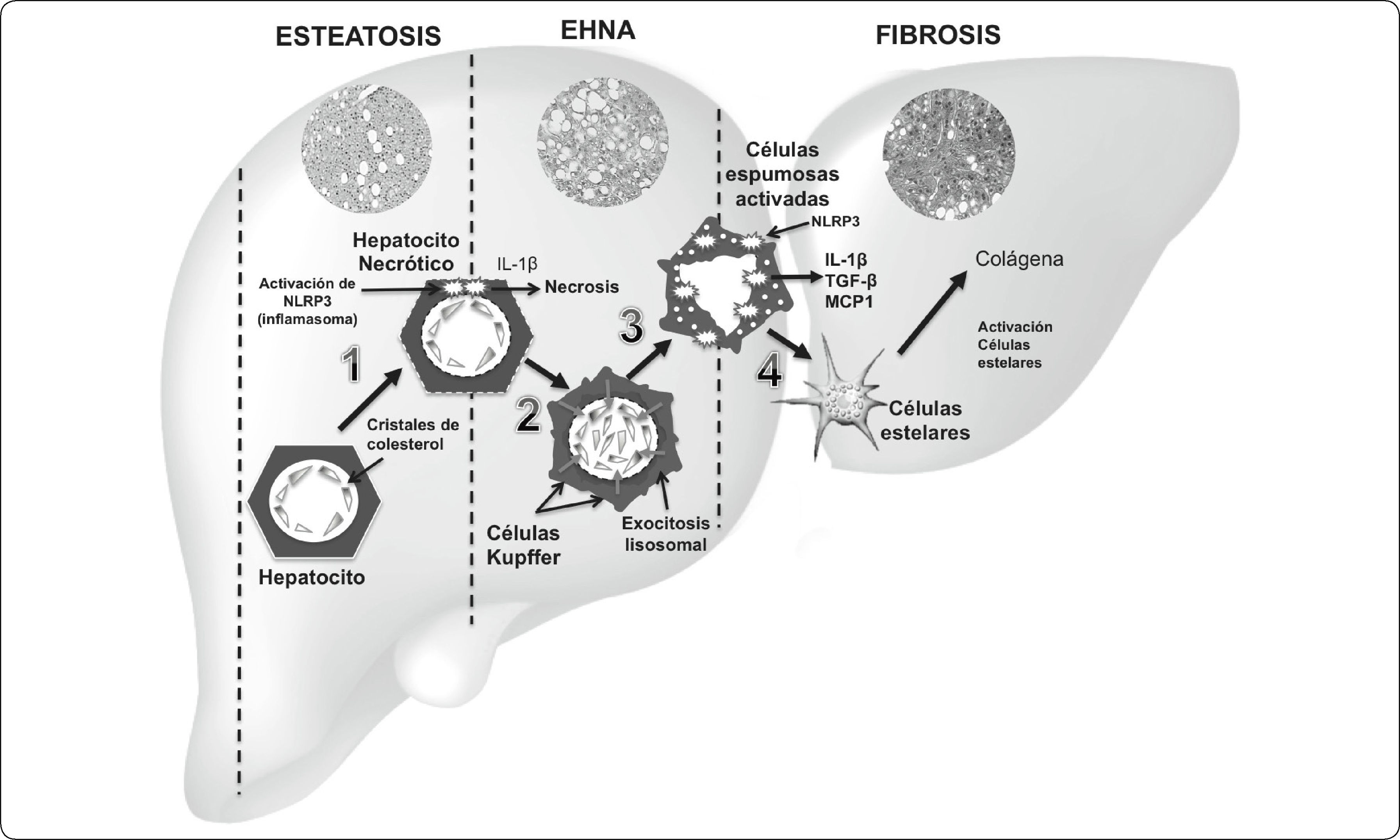
Cristales de colesterol y EHNAA los cristales de colesterol se les ha implicado en la activación del inflamasoma en lesiones arterioescleróticas128,129; por tanto, se ha sugerido que este proceso podría ocurrir de manera similar en EHNA12,60. Por ejemplo, tanto en un modelo de ratón con dieta alta en colesterol, como en sujetos con EHNA, se ha observado que los cristales de colesterol están presentes en las gotas de lípidos, y forman estructuras tipo corona, que conduce a la producción de citocinas pro-inflamatorias, y activación de CKs y CEH 130,131. Recientemente, se ha propuesto un modelo que describe el mecanismo de los cristales de colesterol en el desarrollo a EHNA12: 1) La cristalización del colesterol ocurre en los hepatocitos esteatóticos inicialmente en la periferia de las gotas de lípidos largas, lo cual activa el inflamasoma NLRP3 en los hepatocitos, en consecuencia aumenta la producción de quimiocinas y citocinas pro-inflamatorias (IL-1β, interleucina 1 beta). 2) En respuesta a señales quimiostáticas producidas por los hepatocitos, las CKs se agregan alrededor de los hepatocitos necróticos, que contienen cristales de colesterol, y forman estructuras tipo corona. 3) La exposición de las CKs a los cristales de colesterol causa activación del inflamasoma NLRP3 dentro de las CKs, lo cual conduce a la producción de citocinas pro-inflamatorias y quimiocinas, y en consecuencia se promueve inflamación crónica característica de la EHNA; las CKs expuestas a CL se transforman a células espumosas activadas cargadas de lípidos. 4) Las señales quimiostáticas producidas por la activación de las CKs atrae infiltrado inflamatorio adicional de otras CKs y neutrófilos, así como agregación, activación y transformación de las células estelares a miofibroblastos productores de colágena, lo cual conduce a fibrosis, y en el último de los casos cirrosis12,130,131 (Figura 3).
 Cristalización de colesterol en hepatocitos, activación del inflamasoma NLRP3 y producción de quimiocinas y citocinas. 2) Formación de estructuras tipo corona (ETC) a través de la agregación de células de Kupffer (CKs). 3) Transformación de las CKs a células espumosas activadas por acumulación de cristales de colesterol. 4) Activación y transformación de células estelares hepáticas a miofibroblastos productores de colágena. Abreviaturas: IL-1β, interleucina 1 beta; IL18, Interleucina 18; MCP1, proteína quimioatrayente de monocitos 1; TGF-β, factor de crecimiento transformante beta; NLRP3, inflamasoma que contiene los dominios: LRR (rico en repeticiones de leucina), NOD (domino central de unión a nucleótidos NACHT) y un dominio N-terminal PYD (dominios pirina). Basado en 12,130.")
Modelo de la participación de los cristales de colesterol en el desarrollo de EHNA y fibrosis. 1) Cristalización de colesterol en hepatocitos, activación del inflamasoma NLRP3 y producción de quimiocinas y citocinas. 2) Formación de estructuras tipo corona (ETC) a través de la agregación de células de Kupffer (CKs). 3) Transformación de las CKs a células espumosas activadas por acumulación de cristales de colesterol. 4) Activación y transformación de células estelares hepáticas a miofibroblastos productores de colágena. Abreviaturas: IL-1β, interleucina 1 beta; IL18, Interleucina 18; MCP1, proteína quimioatrayente de monocitos 1; TGF-β, factor de crecimiento transformante beta; NLRP3, inflamasoma que contiene los dominios: LRR (rico en repeticiones de leucina), NOD (domino central de unión a nucleótidos NACHT) y un dominio N-terminal PYD (dominios pirina). Basado en 12,130.
El HGNA es la forma más común de enfermedad hepática crónica. Además, la EHNA podría llegar a ser la causa líder de cirrosis y hepatocarcinoma en los siguientes 10 a 20 años10. Sin embargo, el conocimiento actual de los mecanismos involucrados en la progresión a EHNA y fibrosis, son escasos. Por lo que un mejor entendimiento de la patogénesis del HGNA podría ayudar a direccionar las propuestas terapéuticas para el tratamiento de la EHNA29,60.
La asociación entre el alto consumo de colesterol, así como niveles incrementados de CL y alteraciones en la homeostasis del colesterol hepático, en sujetos con EHNA, ha conducido al estudio de los mecanismos moleculares involucrados en la lipotoxicidad hepática del colesterol. En este sentido, se ha observado que el CL puede causar disfunción mitocondrial y del RE, así como activar CKs y CEHs e inducir inflamación y fibrosis, condiciones que caracterizan a la EHNA. Por lo que actualmente, se ha considerado al CL como una molécula clave en la progresión de la enfermedad.
Llama la atención que aunque distintos mecanismos moleculares en la lipotoxicidad del colesterol se han descrito principalmente en modelos experimentales, aún hace falta evaluar si contribuyen a la patogénesis de la EHNA en humanos; así como determinar, si los genes que participan en la homeostasis del colesterol, podrían ser blancos terapéuticos en la prevención y/o tratamiento de la EHNA y fibrosis.