



La epilepsia es un trastorno neurológico que afecta a 50 millones de personas en el mundo. Se define por la presencia de crisis epilépticas espontáneas resultado de descargas sincrónicas de una población neuronal debido a un dinamismo anormal de las redes neuronales. Diferentes factores han sido implicados en su etiopatogenia, uno de ellos siendo los procesos inmunológicos e inflamatorios. En el presente trabajo revisaremos los datos existentes sobre el papel de la inflamación/neuroinflamación en la epilepsia.
Epilepsy is a neurological disorder affecting 50 million people worldwide. It is defined by the presence of spontaneous seizures result of synchronous discharges of neuronal population due to abnormal dynamics of neural networks. Different factors have been implicated in its pathogenesis, one of them being immune and inflammatory processes. In this paper we review the existing data on the role of inflammation / neuroinflammation in epilepsy.
La epilepsia es un trastorno neurológico que afecta a 50 millones de personas en el mundo; entre el 75 y 80% de los pacientes afectados se encuentran en países en vías de desarrollo. La incidencia de esta enfermedad es de 50/100,000 personas en y 100-190/100,000 personas en países con un índice de crecimiento bajo (Organización Mundial de la Salud, Nota descriptiva N° 999, Mayo 2015; http://www.who.int/mediacentre/factsheets/fs999/es/). La principal característica de este trastorno neurológico es la presencia de crisis epilépticas espontáneas resultado de descargas sincrónicas de una población neuronal debido a un dinamismo anormal de las redes neuronales. Las crisis epilépticas se clasifican en parciales y generalizadas; la principal diferencia entre ellas es que las crisis parciales se originan en una población neuronal y no se expanden a todo el sistema nervioso central, mientras que las generalizadas sí lo hacen1.
Recientemente la Liga Internacional contra la Epilepsia (ILAE) definió a la epilepsia, fármacorresistente y persistente a pesar de haber probado dos regímenes terapéuticos (apropiados y bien tolerados), ya sea como monoterapia o en combinación2. En estos casos, la cirugía de resección del foco epiléptico es una de las opciones terapéuticas, logrando la ausencia de recidiva de crisis en alrededor del 65% de los pacientes3.
La epilepsia del lóbulo temporal (ELT) es la forma más común de epilepsia parcial. Como su nombre lo indica se origina en las estructuras del lóbulo temporal (particularmente en el hipocampo, giro parahipocampal y amígdala) y es fármacorresistente en alrededor del 30% de los pacientes afectados4,5.
La epilepsia es un padecimiento con una etiología multifactorial. En particular, en los últimos años, la relevancia de los procesos inmunológicos e inflamatorios en su etiopatogenia ha sido destacada6–8. Durante décadas, se mantuvo la creencia de que las neuronas epilépticas eran las iniciadoras de las convulsiones. Sin embargo, evidencias recientes han puesto de manifiesto la importancia de la relación entre las células cerebrales (neuronas pero también células gliales) y las células inmunes periféricas9.
En el presente trabajo revisaremos datos recientes sobre el papel de la inflamación/neuroinflamación en la epilepsia.
EVIDENCIAS QUE DEMUESTRAN EL VÍNCULO EPILEPSIA-INFLAMACIÓN: EPILEPSIAS AUTOINMUNESDiversas observaciones realizadas hace algunas décadas han sugerido la implicación de factores inmunoinflamatorios en la epilepsia. La relevancia de la inflamación cerebral en la epilepsia fue inicialmente identificada por estudios patológicos realizados en sujetos afectados por encefalitis de Rasmussen, un síndrome epiléptico caracterizado por pérdida neuronal, inflamación cortical y gliosis confinadas a un hemisferio cerebral10. En estos pacientes se describen en estudios patológicos la presencia de autoanticuerpos de linfocitos B y T citotóxicos, activación de microglia y astrocitos, células Natural Killer (NK), hechos que demuestran la implicación de fenómenos inmunológicos en su patogenia11–17 (Tabla I).
Epilepsias autoinmunes asociadas a la presencia de autoanticuerpos
| Tipo de epilepsia | Auto anticuerpos | Infiltrado inflamatorio | Referencias |
|---|---|---|---|
| Encefalitis deRasmussen | Ac anti-GluR3Ac anti α7 nACh receptorAc anti-Munc18-1 | Linfocitos T CD8 | Rogers et al., 199433Watson et al., 200511Álvarez Barón et al., 200812 |
| Encefalitis límbica | Ac-anti AMPARAc-anti GABAbRAc-anti NMDARAc-anti VGKC (LGI1, CASPR2, Contactin-2)Ac-anti GAD | Linfocitos BLinfocitos T CD4Microglia activada | Vincent et al., 2011 a,b14,20Quek et al., 201215Bien et al., 201216Granata et al., 201113 |
| Encefalitis paraneoplásica límbica | Ac antineuronal nuclear tipo 1.Ac-anti CRMP-5Ac-anti HuAc-anti Ma2Ac-anti AbamphiphysinAc anti CV2Ac-anti Sox1 | Linfocitos T CD8/CD3NK CD57+Linfocitos B/ Células plasmáticas (CD138+)Astrocitos activadosMicroglia activada | Quek et al., 201215Bien CG et al., 201216 |
| Síndrome Landue-Kleffner | Ac anti-BDNFAc anti-células endoteliales | Hirsch et al., 200634 | |
| Enfermedad de Batten | Ac-anti GAD65 | Astrocitos activados | Granata et al., 201113Chattopadhyay et al., 200235 |
Así mismo la eficacia de la hormona Adrenocorticotrófica (ACTH) y de los corticosteroides utilizados en diferentes epilepsias pediátricas, conocidas ya desde hace décadas, hace sugerir también un vínculo inflamación/epilepsia, como ha sido demostrado recientemente18.
La relación entre la activación del sistema inmune y la presencia de crisis fue posteriormente consolidada por la identificación de auto- anticuerpos circulantes en ciertos pacientes con epilepsia (Tabla I), así como por la alta frecuencia de epilepsia en diferentes enfermedades autoinmunes13,19,20.
EVIDENCIAS PATOLÓGICAS: EXISTE UN FENÓMENO NEUROINFLAMATORIO ASOCIADO A LA EPILEPSIA.Estudios histológicos realizados en el tejido cerebral de pacientes con epilepsia a priori no vinculadas al fenómeno inflamatorio, han demostrado la inducción de varias vías de señalización inflamatorias, la sobrexpresión de genes de quimiocinas y citocinas pro-inflamatorias, así como una prominente activación de la microglia y de los astrocitos21–27. La presencia de células inmunes periféricas en el tejido cerebral y la activación de la vía de señalización de receptores tipo Toll (TLR) han sido demostradas recientemente28,29. Estudios de tomografía de emisión de positrones (PET) con el marcador C-PK11195 (específico de microglia activada), confirmaron estos hallazgos en pacientes con epilepsia secundaria a encefalitis30,31 o con displasia cortical focal32.
Estas observaciones permitieron establecer que el fenómeno inmuno-inflamatorio está asociado intrínsecamente a la epilepsia, independientemente de su etiología.
LAS CRISIS EPILÉPTICAS INDUCEN NEUROINFLAMACIÓN Y VICEVERSALa relación de causalidad entre epilepsia e inflamación se ha podido explorar en condiciones controladas y con mayor profundidad en modelos experimentales. Su uso ha permitido demostrar que las crisis recurrentes inducen neuroinflamación con activación de microglia, de astrocitos, de neuronas, así como de las células epiteliales de la barrera hemato-encefálica6,7,36.
Sin embargo, a su vez la neuroinflamación favorece la ocurrencia de crisis epilépticas, ya que se ha demostrado que la neuroinflamación es un factor que favorece la ocurrencia de crisis y agrava su patología. En particular se sabe que las citocinas proinflamatorias liberadas por la glía, tienen un papel importante en la hiperexcitabilidad neuronal implicada en la generación de crisis epilépticas y su recurrencia, así como en el daño celular excitotóxico asociado. Esta hiperexcitabilidad es favorecida por alteraciones en la regulación mediada por células gliales de los neurotransmisores, iones y de agua. El descontrol de la respuesta inmune mediada por las células gliales puede causar cambios inflamatorios sostenidos los cuales facilitan la epileptogénesis37. Por otro lado, se ha observado que el bloqueo farmacológico o la inactivación de diferentes vías pro-inflamatorias (IL-1, TNF-a, COX-2) o de elementos de las vías de señalamiento del receptor tipo Toll-like resulta en potentes efectos anticonvulsivantes23,38–40. Se sabe que en modelos de ratones que sobre-expresan la forma soluble del receptor antagonista de IL1β (IL-1Ra), en astrocitos son intrínsecamente resistentes a las crisis epilépticas41,42, mientras que ratones carentes del gen ICE/Caspasa-1 o del gen IL-1R (los cuales son incapaces de producir y liberar IL-1β o de activar la señalización de IL-1β, respectivamente), muestran un retraso significativo en el inicio de las crisis epilépticas, presentando resistencia a crisis futuras39,40.
BARRERA HEMATO-ENCEFÁLICA Y EPILEPSIASe ha demostrado que las crisis epilépticas y/o el estatus epiléptico se asocian con un aumento en la permeabilidad de la barrera hemato-encefálica, lo cual pudiese participar en la epileptogénesis y en la progresión de la epilepsia43,44. En efecto, las crisis convulsivas prolongadas conllevan una sobre regulación de las moléculas de adhesión ubicadas en las células endoteliales, facilitando la extravasación de los linfocitos circulantes; en particular, la expresión de moléculas como la E-selectina, P-selectina, ICAM-1 y VCAM-1 se encuentran aumentadas45,46. Los ligandos de estas moléculas son expresados en los leucocitos circulantes después de las crisis lo que facilita su entrada en el sistema nervioso central47. Se ha mostrado en particular que el bloqueo de la α4β1 integrina sobre los leucocitos inhibe su infiltración en el cerebro, y que la inhibición terapéutica de la activación de la α4 integrina previene la inducción de crisis, así como el desarrollo de la epilepsia47. Así, la infiltración de las células inmunes periféricas parece tener un papel relevante en la inducción de las crisis y en el desarrollo de la epilepsia, aunque aún falta información suficiente, para entender a cabalidad el alcance que este hecho pudiese tener9.
INFLAMACIÓN PERIFÉRICA, NEUROINFLAMACIÓN Y EPILEPSIADiferentes estudios clínicos han demostrado la existencia de un incremento en los niveles de mediadores inflamatorios (IL-6, TNF-α, IL-1β, IL-1β Ra, entre otros) en suero y líquido cefalorraquídeo de pacientes con epilepsia48.
Otros estudios apoyan el hecho de que el estado inflamatorio periférico puede influenciar la presencia de las crisis epilépticas. Se ha mostrado que la inflamación sistémica disminuye el umbral de las crisis epilépticas en varios modelos experimentales de inflamación periférica49–52. En particular, en un modelo de colitis inflamatoria, la intensidad de las crisis se correlacionó significativamente con la severidad de la inflamación periférica y fue revertida con la resolución natural del fenómeno inflamatorio53. Así mismo se demostró también la presencia de una respuesta inflamatoria cerebral durante la inflamación periférica, ya que fueron detectados niveles altos de TNF-α y de activación microglial en el hipocampo, con lo cual queda de manifiesto que la inflamación periférica aumenta la excitabilidad del SNC en las personas con epilepsia.
Debido a la presencia de infiltración de células inmunes periféricas en el SNC durante las crisis epilépticas, el conocer la composición celular periférica en pacientes epilépticos pareciera muy relevante. Comparando el fenotipo de leucocitos periféricos entre pacientes con epilepsia en periodo interictal (tiempo trancurrido entre una crisis y otra), se encontró un elevado porcentaje de monocitos y de células NK en todos los pacientes incluidos, así como una disminución de los linfocitos B en los pacientes con epilepsia focal, ambos hallazgos comparando con los controles sanos54.
Así mismo, se encontró que niños con síndrome de West (epilepsia caracterizada por espasmos tónicos breves asociados a un aspecto electroencefalográfico particular denominado hipsarritmia y alteraciones en el desarrollo psicomotor), antes del tratamiento con ACTH presentaban un fenotipo linfocitario periférico particular, comparando con sujetos controles, caracterizado por una disminución significativa en los linfocitos CD3+CD25+, CD19+ y CD19+CD95+55. De igual forma, comparando niños con epilepsia contra controles sanos, se observó una disminución significativa de los porcentajes de las CD4+CD25+FoxP3 y CD4+, así como un aumento significativo de las CD8+, NK y células B en los pacientes con epilepsia56. Esta observación sugiere la relevancia del estado inmune periférico en la fisiopatología de esta enfermedad aunque los mecanismos involucrados quedan todavía desconocidos.
INFLAMACIÓN PERIFÉRICA, NEUROINFLAMACIÓN Y EPILEPSIA FÁRMACORRESISTENTELa epilepsia fármacorresistente se asocia a una mortalidad cinco veces más alta en comparación con la población en general57. Se define una epilepsia como fármacorresistente cuando se han ensayado uno o dos medicamentos en dosis terapéuticas diarias sin que se logre un control de las crisis convulsivas. Las causas de la epilepsia fármacorresistentes son numerosas, muchas de ellas pueden ser debido a anormalidades en la maduración cerebral, a lesiones cerebrales graves con cambios irreversibles respecto a la organización de la neuroglia cerebral y a una función inhibitoria neuronal, entre otras.
Algunas de las estrategias que se han tomado para tratar esta condición, incluyen la resensibilización de las neuronas hacia cierto tipo de drogas mediante métodos moleculares, la inhibición de la sobreexpresión de transportadores de proteínas mediante la utilización de bloqueadores de canales de calcio (Verapamil), que inducen un incremento en la concentración intracelular de las drogas antiepilépticas4, la utilización de la atorvastatina que es un inhibidor de la muerte celular hipocampal inducida por un agonista del ácido kaínico produciendo neuroprotección y acción antiepiléptica58 y la estimulación eléctrica del nervio vago (EENV).
El nervio vago es el décimo nervio craneal y es el principal componente del sistema parasimpático del sistema nervioso autónomo. Su principal neurotransmisor es la acetilcolina. Se ha reportado que la EENV es efectiva en diferentes tipos de crisis epilépticas parciales y recientemente en crisis epilépticas generalizadas (reducción entre 30 y 85%). Este efecto ha sido demostrado tanto en la población infantil como adulta59. En modelos experimentales de epilepsia se ha también demostrado la efectividad de la EENV. Parámetros como el número de crisis, la severidad de la crisis, la frecuencia de las crisis, la latencia de la primera crisis, etc; han sido disminuidos como consecuencia de la EENV60. En un modelo de displasia cortical focal, la cual produce crisis epilépticas la EENV fue capaz de abolir la actividad epiléptica61. Sin embargo, hasta el momento, el mecanismo preciso para la efectividad de la estimulación eléctrica del nervio vago en el control de las crisis, es aún desconocido.
Hasta el momento lo que se sabe es que la EENV disminuye la inflamación periférica. Se ha demostrado en un modelo de inflamación periférica inducido por la administración de un lipopolisacárido, que la EENV disminuye la concentración de TNF-α tanto en suero como en hígado62. En un modelo experimental de golpe de calor el cual genera inflamación sistémica, también se ha mostrado el mismo efecto; la EENV permite disminuir la concentración de citocinas pro-inflamatorias como TNFα e IL-6 en el pulmón a diferentes tiempos63.
Como hemos mencionado anteriormente el estado inflamatorio periférico es capaz de influenciar el estado inflamatorio del sistema nervioso central. Y se sabe que la EENV es eficaz en el control de crisis epilépticas, así como en el control de la inflamación sistémica. Así podría ser factible que uno de los mecanismos que participe en la eficiencia de la EENV en las epilepsias fármacorresistentes sea la modulación del estadio inflamatorio periférico.
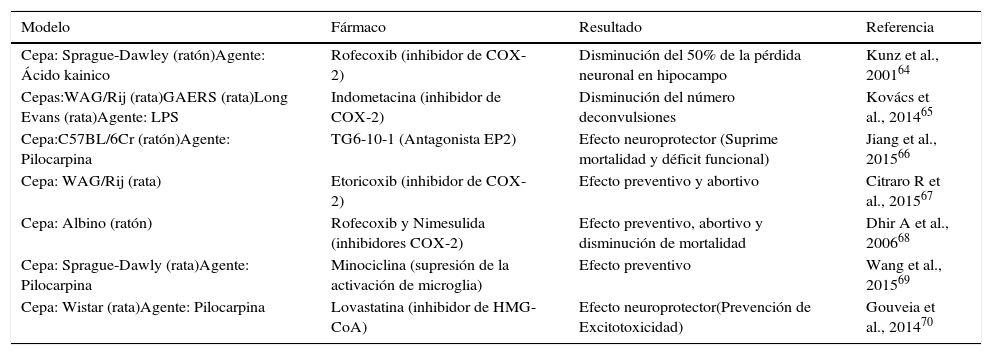
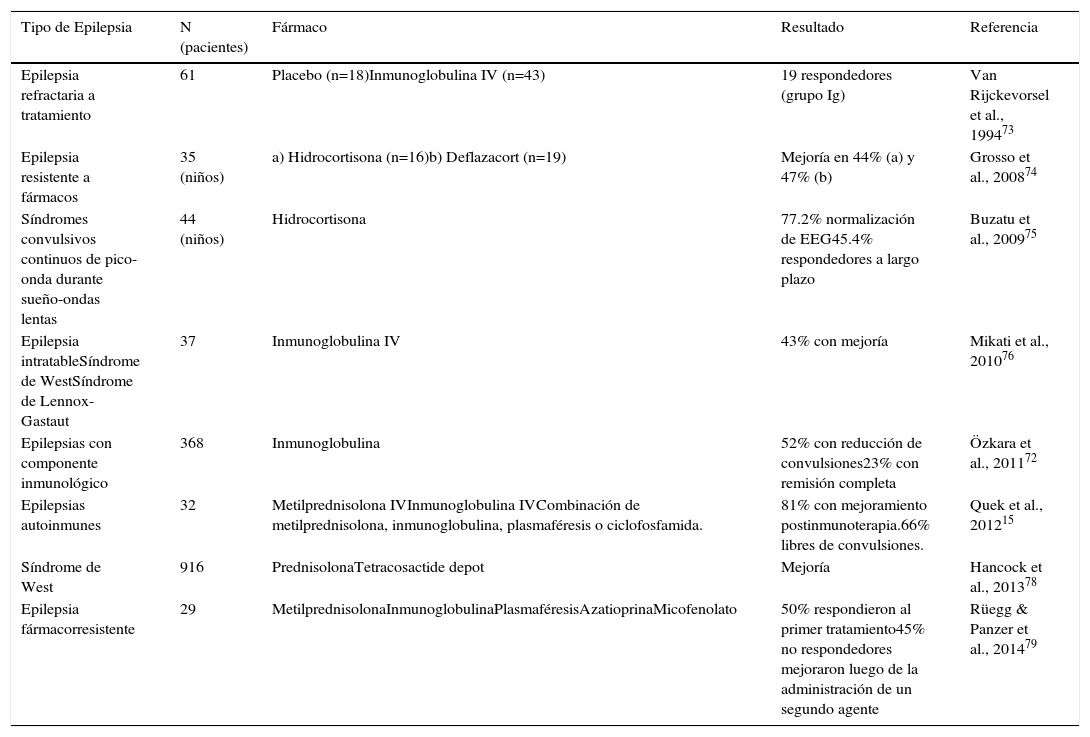
TRATAMIENTOS ANTIINFLAMATORIOS/INMUNOMODULADORES Y EPILEPSIAComo se mencionó anteriormente, los tratamientos anti-inflamatorios y/o inmunomoduladores, han mostrado ser útiles desde hace tiempo en pacientes con epilepsia con un claro componente inmunológico71,72. Considerando estas evidencias, se han explorado en diferentes modelos experimentales el potencial de tratamientos antiinflamatorios sobre la epileptogénesis y las crisis15,72–79 (ver Tabla II). En particular, se ha mostrado que la inhibición de COX-2 tiene efecto antiepileptogénico67,80 y que la minociclina (antibiótico con potentes efectos antiinflamatorios), permite reducir la frecuencia, duración y severidad de las crisis epilépticas en un modelo de epilepsia del lóbulo temporal69. Si bien los estudios en pacientes, son hasta el momento escasos se han reportado algunos protocolos que se encuentran actualmente en curso72 (Tabla III). Uno de los problemas es lograr elegir los pacientes que podrían beneficiarse de una terapia antiinflamatoria asociada48. Así, el contar con biomarcadores periféricos de neuroinflamación podría, en un futuro, permitir mejorar las indicaciones del tratamiento médico de estos pacientes48,81.
Tratamientos antiinflamatorios/inmunomoduladores en epilepsia: Evidencias experimentales
| Modelo | Fármaco | Resultado | Referencia |
|---|---|---|---|
| Cepa: Sprague-Dawley (ratón)Agente: Ácido kainico | Rofecoxib (inhibidor de COX-2) | Disminución del 50% de la pérdida neuronal en hipocampo | Kunz et al., 200164 |
| Cepas:WAG/Rij (rata)GAERS (rata)Long Evans (rata)Agente: LPS | Indometacina (inhibidor de COX-2) | Disminución del número deconvulsiones | Kovács et al., 201465 |
| Cepa:C57BL/6Cr (ratón)Agente: Pilocarpina | TG6-10-1 (Antagonista EP2) | Efecto neuroprotector (Suprime mortalidad y déficit funcional) | Jiang et al., 201566 |
| Cepa: WAG/Rij (rata) | Etoricoxib (inhibidor de COX-2) | Efecto preventivo y abortivo | Citraro R et al., 201567 |
| Cepa: Albino (ratón) | Rofecoxib y Nimesulida (inhibidores COX-2) | Efecto preventivo, abortivo y disminución de mortalidad | Dhir A et al., 200668 |
| Cepa: Sprague-Dawly (rata)Agente: Pilocarpina | Minociclina (supresión de la activación de microglia) | Efecto preventivo | Wang et al., 201569 |
| Cepa: Wistar (rata)Agente: Pilocarpina | Lovastatina (inhibidor de HMG-CoA) | Efecto neuroprotector(Prevención de Excitotoxicidad) | Gouveia et al., 201470 |
Tratamientos antiinflamatorios/inmunomoduladores en epilepsia: Evidencias clínicas
| Tipo de Epilepsia | N (pacientes) | Fármaco | Resultado | Referencia |
|---|---|---|---|---|
| Epilepsia refractaria a tratamiento | 61 | Placebo (n=18)Inmunoglobulina IV (n=43) | 19 respondedores (grupo Ig) | Van Rijckevorsel et al., 199473 |
| Epilepsia resistente a fármacos | 35 (niños) | a) Hidrocortisona (n=16)b) Deflazacort (n=19) | Mejoría en 44% (a) y 47% (b) | Grosso et al., 200874 |
| Síndromes convulsivos continuos de pico-onda durante sueño-ondas lentas | 44 (niños) | Hidrocortisona | 77.2% normalización de EEG45.4% respondedores a largo plazo | Buzatu et al., 200975 |
| Epilepsia intratableSíndrome de WestSíndrome de Lennox-Gastaut | 37 | Inmunoglobulina IV | 43% con mejoría | Mikati et al., 201076 |
| Epilepsias con componente inmunológico | 368 | Inmunoglobulina | 52% con reducción de convulsiones23% con remisión completa | Özkara et al., 201172 |
| Epilepsias autoinmunes | 32 | Metilprednisolona IVInmunoglobulina IVCombinación de metilprednisolona, inmunoglobulina, plasmaféresis o ciclofosfamida. | 81% con mejoramiento postinmunoterapia.66% libres de convulsiones. | Quek et al., 201215 |
| Síndrome de West | 916 | PrednisolonaTetracosactide depot | Mejoría | Hancock et al., 201378 |
| Epilepsia fármacorresistente | 29 | MetilprednisolonaInmunoglobulinaPlasmaféresisAzatioprinaMicofenolato | 50% respondieron al primer tratamiento45% no respondedores mejoraron luego de la administración de un segundo agente | Rüegg & Panzer et al., 201479 |
La epilepsia es uno de los padecimientos neurológicos más prevalentes, causante de una morbilidad y mortalidad elevadas. Aunque existen varios tratamientos eficientes, las epilepsias fármacorresistentes siguen siendo frecuentes. La relevancia de la inflamación central y probablemente periférica en su patogenia es un hallazgo muy interesante, ya que la modulación de éstos podría permitir mejorar el pronóstico de este padecimiento.
Este trabajo se realizó con el financiamiento parcial del CONACYT (Proyecto Salud-2013-1-201448).