


Ste2p is a G protein-coupled receptor (GPCR) in Saccharomyces cerevisiae that mediates mating by responding to the alpha-mating factor pheromone. Ste2p belongs to a subfamily of GPCRs with no global sequence similarity to GPCRs of known atomic three-dimensional structure, yet it shares functional similarities with many of these. To deepen our understanding of the structure-function relationship of this receptor, we built an atomic three-dimensional homology-based model of Ste2p that was used to simulate the docking of the alpha pheromone. The Ste2p model is in general agreement with the available experimental data and allowed us to propose that the interface between Ste2p and alpha pheromone is formed by 26 residues, most of which are polar residues located at the three extracellular loops and helices HI, H5, and H6. This interface does not include Ile190, a highly conserved residue among fungal species, located at the second extracellular loop and therefore a potential binding site residue. By performing mutagenesis of STE2 at this position we observed only a small effect of this residue in receptor signaling. Hence, the Ste2p model presented here is consistent in general with current experimental data and constitutes a framework to test hypothesis about the structure-function relationship of this receptor.
Ste2p es un receptor acoplado a la proteína G (GPCR) en Saccharomyces cerevisiae que se une a la feromona alfa para mediar el apareamiento. Ste2p pertenece a una subfamilia de GPCRs que no presentan homología global en secuencia con los GPCRs de estructura atómica tridimensional conocida, pero comparte propiedades funcionales con muchos de éstos. Para profundizar nuestro entendimiento de la relación estructura-función de este receptor, en este trabajo presentamos un modelo de la estructura atómica tridimensional de Ste2p asociado a su ligando. El modelo de Ste2p generado es congruente con la información experimental disponible y sugiere que la interfaz entre Ste2p y la feromona está compuesta por 26 residuos, en su mayor parte polares, localizados en las tres asas extracelulares y las hélices H1, H5 y H6. La interfaz no incluye a la Ile190, un residuo altamente conservado entre especies de hongos, que se encuentra en el asa extracelular 2 y es un potencial sitio de anclaje. Mutantes en esta posición en STE2 muestran un efecto pequeño en la señalización del receptor. El modelo presentado de Ste2p es consistente en general con los datos de mutagénesis disponibles a la fecha, por lo que constituye un marco de referencia para evaluar hipótesis sobre la relación estructura-función en este receptor.
Over the past four decades extensive research has been carried out about the structure-function relationships of G protein coupled receptors (GPCRs), promoted by both basic and applied aspects of this group of receptors; more than 40% of drugs in clinical use target GPCRs1 and many fundamental aspects of cell physiology are regulated by these receptors2. Relevant to this study, the GPCR Ste2p from Saccharomyces cerevisiae mediates mating by recognizing the tridecapeptide mating factor, alpha pheromone.
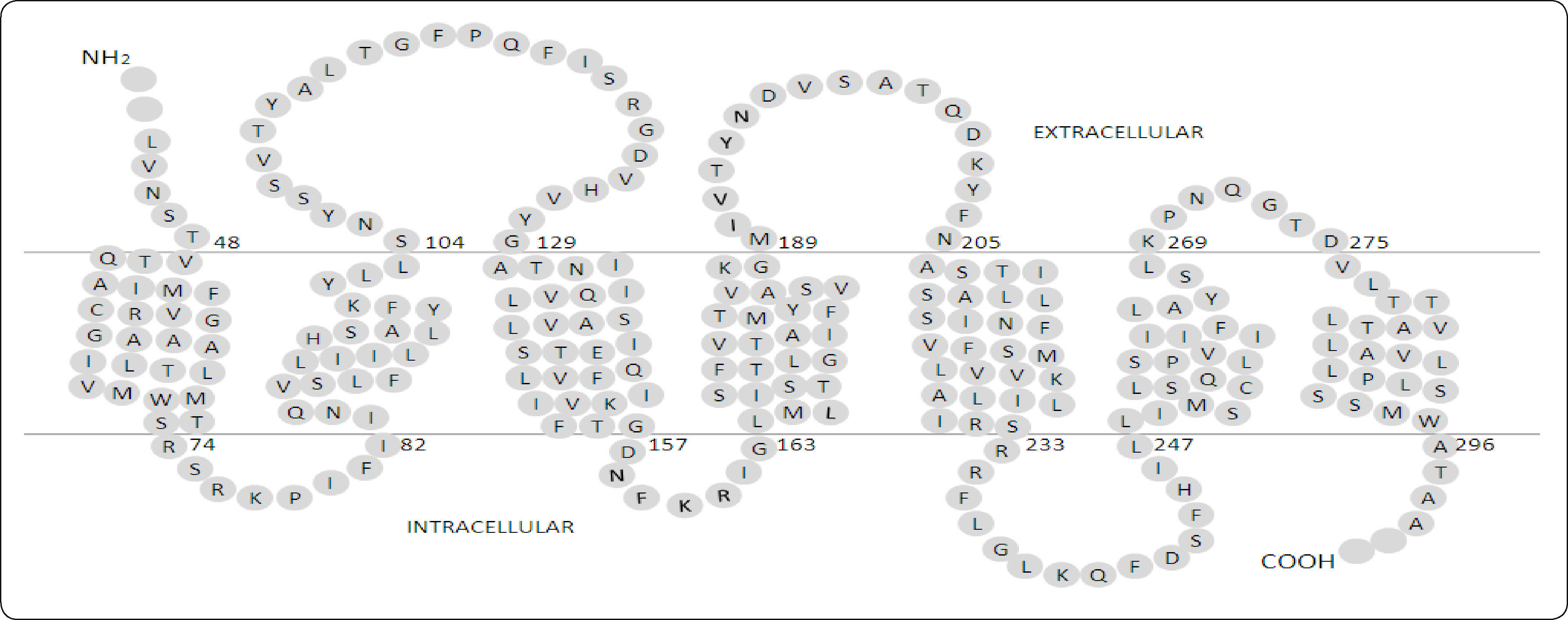
The first studies on Ste2p were reported in 1970-1980 when the molecular basis of mating in yeast cells was discovered. Many genes rendering a sterile (STE) phenotype were discovered, and among these was STE23,4 that was further cloned and characterized5,6. A few years later, the topological structure of Ste2p was inferred through solvent accessibility studies (Figure 1). Due to difficulties to purify and crystallize integral membrane proteins, the atomic three-dimensional structure of Ste2p has not yet been determined. However, NMR was used to determine the structures of two fragments of this receptor: the HI-H2 segment (Protein Data Base entry code: 2K9P), and a segment of H6 (PDB entry code: 1PJD)7,8.

Topological diagram of Ste2p. Every amino acid residue from Ste2p is represented by a single letter code within a gray circle. The two horizontal lines separating the extracellular from the intracellular spaces represent the membrane. Data derived from20.
Several residues and regions that are critical for STE2 function have been determined by site-directed mutagenesis. For example, the N-terminal domain mediates oligomerization of the receptor9,10 while the C-terminal domain acts as negative regulator11–13. The third cytoplasmic loop interacts with the trimeric G protein14, and the first extracellular loop undergoes a conformational change upon ligand binding15 and also plays a role in signal transduction16.
Three different 2D models of Ste2p bound to the pheromone have been proposed and were built based on biochemical data derived until 2004 in an attempt to recapitulate the structure-function studies of this receptor. Briefly, Lee B. K. and colleagues17 proposed a model where both the N and C-termini of the pheromone are buried inside the transmembrane helices of Ste2p, whereas the central region, including a turn structure, interacts with the extracellular domains. In that model, two residues of the receptor, Ser47 and Thr48, were proposed to interact with Gln10 of the pheromone. Lin J. C. and colleagues18 proposed a model that highlights the role of aromatic residues in the interaction between the pheromone and the pocket formed by the extracellular ends of the transmembrane helices in the receptor. In this model, a β-bend is formed at the central region of the pheromone. Tyr266 on helix 6 is oriented toward the surrounding lipids and interacts with Trp1-His2-Trp3 of the pheromone, while Phe204, located within the helix bundle between helix 4 and helix 5, interacts with Tyr13 of the pheromone. Finally, Son C. D. and colleagues19 proposed a model where Gln10 of the pheromone interacts with residues Ser47 and Thr48 in Ste2. Tyr13 in the pheromone interacts with helix 1 (Phe55−Arg58) and Trp1 and Trp3 in the pheromone interact with aromatic residues Phe262 and Tyr266 at the extracellular interface of helix 6. All these models have in common that the central region of the pheromone forms a turn structure that is oriented away from the transmembranal helix-bundle, whereas both the N and C-termini of the pheromone are oriented toward the binding pocket formed by extracellular ends of helix 1, helix 5, and helix 6.
In the present study, we built a novel three-dimensional model of Ste2 bound to the pheromone, incorporating mutagenesis information about Ste2p function that was not necessarily incorporated in previous models of this interaction; our model was built using the state of art tools used to build three-dimensional models of proteins. In our model, Ile190 in Ste2p, a conserved position among fungal species, does not interact with the pheromone. To test our model, we performed a mutagenesis at this residue. We choose this residue because it is part of the second extracellular loop, which has been previously implied to play a role in ligand recognition and binding in other GPCRs, such as the dopamine D221, MT2 melatonin22, thyrotropin23 angiotensin24, and histamine H125 receptors. Our results indicate that some mutants of this residue have a small effect on the Ste2p signaling cascade that leads cells to arrest cell growth in the presence of the pheromone. The structural bases of these results are discussed using our three-dimensional model.
MATERIAL AND METHODSThe construction of a Ste2 model was done using the rhodopsin crystal as a template. The receptor model was built using the i-TASSER V4.1 server26. i-TASSER was chosen because it was ranked as the server with best results in CASP7 to CASP10 (Critical Assessment of Techniques for Protein Structure Prediction). We modeled residues 47 to 296. The N-terminal (residues 1-46) and C-terminal domains (residues 297-431) were not modeled because these domains are not essential for ligand binding.
Despite the fact that Ste2p and rhodopsin have a low sequence identity (20%), rhodopsin was used as a template for Ste2p modeling because: 1) Ste2p and rhodopsin share structural and functional properties, 2) available structural and mutational data show that the Ste2p and rhodopsin sequences align properly based on a comparison of amino acids that have similar structural and functional roles in membrane proteins27 and 3) rhodopsin has been used as a template for previously developed Ste2p models18. The sequences for Ste2p and rhodopsin were obtained from the UniProt database. The crystal structure of rhodopsin (PDB entry code 1U1928) was obtained from the Protein Data Bank29. The alignment of Ste2p with rhodopsin was carried out as described previously27.
The membrane boundaries in the Ste2p sequence were obtained from solvent accessibility data20 and were used in i-TASSER to specify boundaries of secondary structures. To guide i-TASSER on the correct helix orientation, distance restraints between specific residues were obtained from the PDB file of rhodopsin using the distance between the alpha-carbon from a residue placed at the end of a helix and the alpha-carbon from the residues localized at the ends of all the other helices.
Construction of the alpha-pheromone three-dimensional model. The PEP-FOLD server30 was used to develop an atomic three-dimensional model of the alpha pheromone peptide. The sequence of alpha pheromone Trp-His-Trp-Leu-Gln-Leu-Lys-Pro-Gly-Gln-Pro-Met-Tyr was sent to the server in FASTA format. The model that was closer to the structural characterization of the pheromone by NMR31 was selected from the results generated by the PEP-FOLD server.
Docking of Ste2 with alpha pheromone. For docking experiments the ClusPro 2.0 server32 was used. Amino acid residues of the Ste2p receptor and the pheromone involved in binding were used to focus the docking on such residues. These residues included (Ste2p-pheromone pairs; the secondary structure element of the Ste2p residue is indicated in parenthesis for convenience): Ser47(H1)-Gln1017, Thr48(H1)-Gln1017, Phe204(H5)-Tyr1318, Asn205(H5)-Trp333, Tyr266(H6)-Trp318 and Lys269(H6)-Trp134. The energy of the Ste2p-alpha pheromone complex was minimized using CHARMM35; for that end the C38B2 CHARMM version and CHARMM36 potentials were used. The complex that showed the lowest interaction energy with the lowest average distance between contacts was selected. All the structures were visually inspected with the PyMol viewer software36.
Site-directed mutagenesis of the STE2 gene. Mutations were introduced into the receptor gene (STE2) by PCR using the kit “QuikChange Lightning” (Agilent Technologies Cat. No. 210519).
The pYES-STE2 plasmid was used as template for PCR and was constructed by subcloning the 1296bp STE2 gene into the pYES2 plasmid (Invitrogen Cat. No. V82520). STE2 was obtained from the pGRB2.2-STE2 plasmid using the XbaI and EcoRI restriction enzymes. The pGRB2.2-STE2 plasmid was kindly provided by Dra. Irene Castaño (Instituto Potosino de Investigación Científica y Tecnológica, San Luis Potosí, México). Mutagenic primers were designed according to manufacturer's instructions and were complementary to the STE2 sequence, except for the AUU codon of Ile190 that was changed for NNG/C to introduce the mutations. Sequences of the primers used are described next:
- 1)
5’GCGCTGTTAAAGGTATGNNG/CGTGACTTATAAT GATGTTAGTGCCACCC 3’
- 2)
5’ GGGTGGCACTAACATCATTATAAGTCACG/CNNCA TACCTTTAACAGCGC 3’.
The whole vector pYES-STE2 was amplified using PfuTurbe polymerase. Then, the PCR product was digested with DpnI restriction enzyme at 37°C for 30minutes to eliminate the parental (non mutated) plasmid, and then transformed into DH5α E. coli strain. Transformed cells were selected on ampicillin-containing plates and plasmids were isolated from transformed cells using QIAprep Spin Miniprep Kit (QIAGEN Cat. No. 27106). Isolated mutants were confirmed by DNA sequence analysis. DNA sequencing was carried out in the Unidad de Biología Molecular, located at the Instituto de Fisiología Celular. Constructs were transformed into a STE2 null mutant MATa strain (BY4741 ΔYFLO26W::kanMX4) as previously described elsewhere37 and transformed cells were selected by their growth in minimal medium lacking uracil.
Growth curves. Yeast cultures expressing the mutant receptor were incubated for 24hours at 30°C with minimal medium lacking uracil with glucose, and then were incubated for 6 additional hours with minimal medium lacking uracil with galactose in order to induce the expression of STE2 in the plasmid. Afterwards, yeast cells were diluted to optical density of 0.11 and incubated with pheromone (Anaspec) at 10μM into a 96-well half area plate in a total volume of 150μl. The incubation was carried out at 30°C with shaking and the A600nm was measured every hour for 24h using a Synergy MX plate reader (BioTek Instruments). The experiment was repeated three times each time with a triplicate. Growth curves data were normalized and the area under the curve was calculated for each mutant.
RESULTS AND DISCUSSIONThree-dimensional model of Ste2p. The Ste2p model with the best C-score (confidence score) was selected (Figure 2); in our case the C-score was -1.01 and the TM-score was 0.30. The C-score given by i-TASSER estimates the quality of predicted models is in the range of [-5, 2]. Models with C-score >-1.5 tend to be close to the native structure. The TM-score provided by i-TASSER estimates the similarity of the protein model and its template; a score below 0.17 corresponds to two proteins that are not similar while above 0.5 correspond to proteins that are structurally homologues. Thus, the TM-score indicates the Ste2p model differs from the rhodopsin template (1U19) as expected (see Methods), but remains similar. In our three-dimensional model the boundaries of the transmembrane helices matched the solvent accessibility data previously described9, except for the H6. This helix has been reported to expand from Leu248 to Leu268 (Figure 1), while in our model it spans from Phe244 to Ile263. This difference reflects the fact that the loop connecting H5 to H6 presents two residues (244 and 245) at the end of this loop that are not accessible to biotinylation, followed by two other residues that are accessible to biotinylation (246 and 247). The authors proposed that H6 should start at residue 248, but we prefer to use the first residues that were inaccessible as part of this helix. These authors could not establish the end of this helix, so we used as a criterion the sequence alignment between Ste2p and rhodopsin generated with i-TASSER.
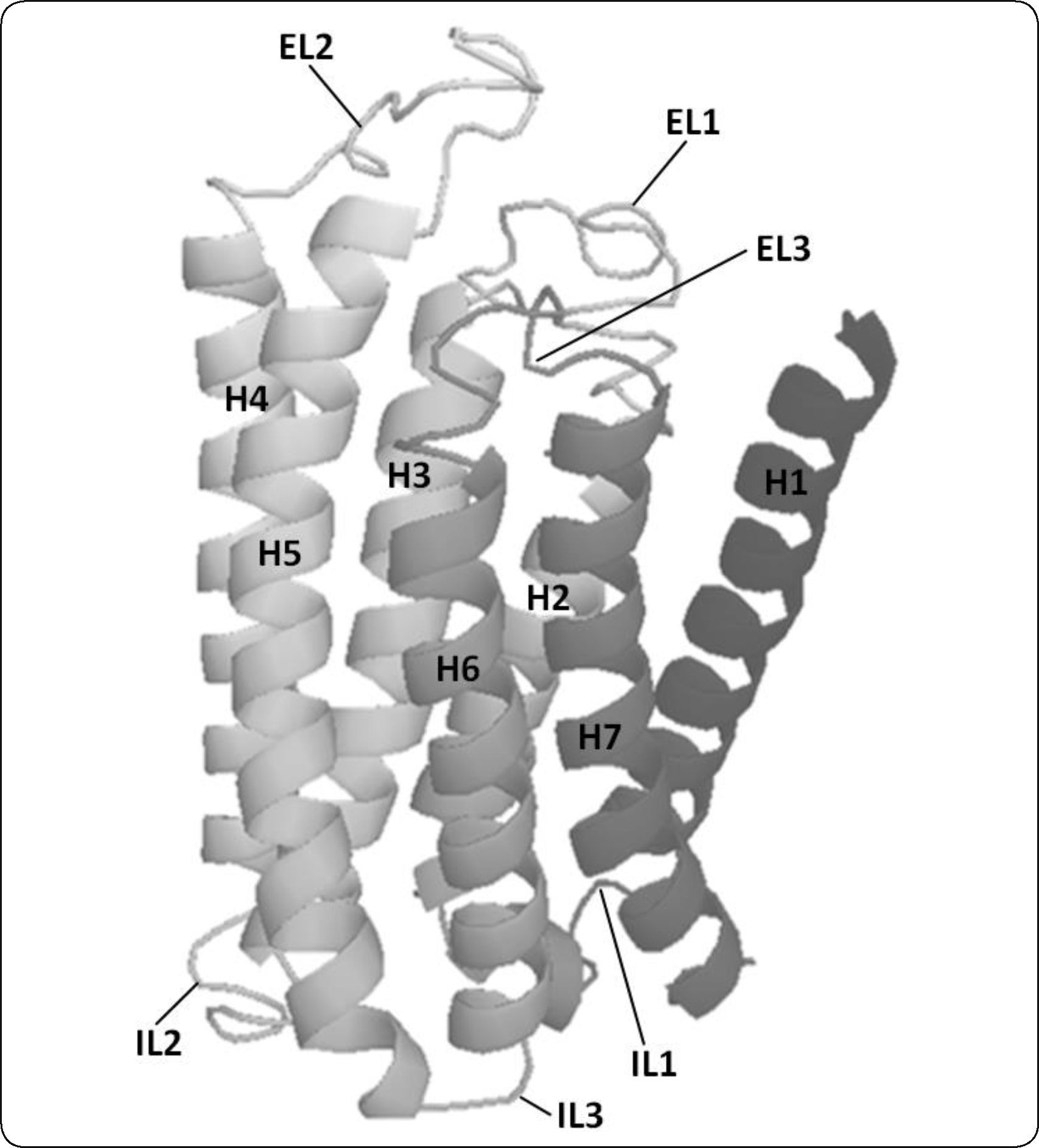
 connected by three intracellular (IL1, IL2 and IL3) and three extracellular loops (EL1, EL2 and EL3). This model shows the counterclockwise orientation of transmembrane helices used for modeling GPCRs. The image was generated with PyMol.")
Three-dimensional structural model of Ste2p. A ribbon representation of the three-dimensional structure of Ste2p is presented. Ste2p has a central core made of seven transmembrane helices (HI to H7) connected by three intracellular (IL1, IL2 and IL3) and three extracellular loops (EL1, EL2 and EL3). This model shows the counterclockwise orientation of transmembrane helices used for modeling GPCRs. The image was generated with PyMol.
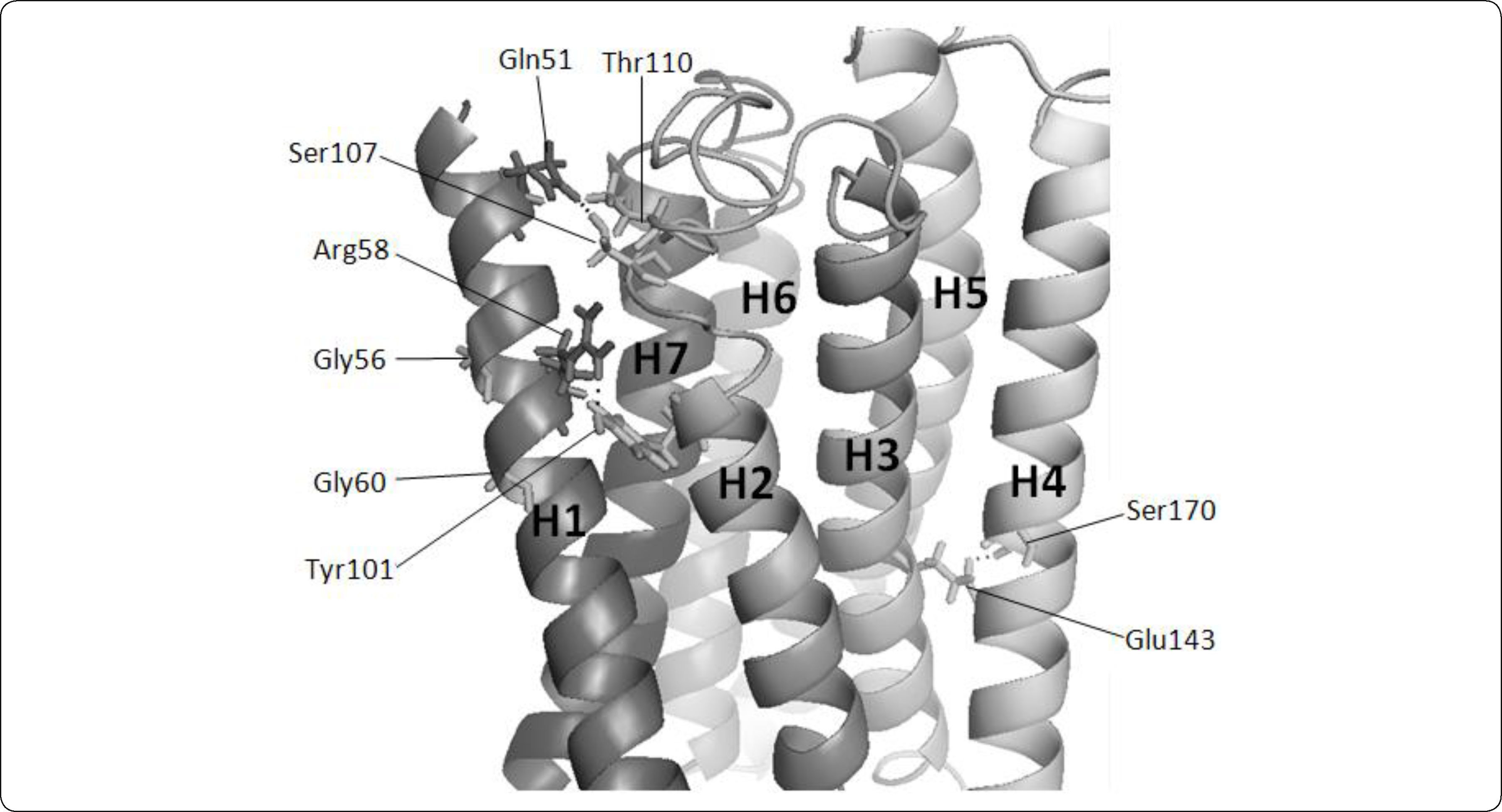
The Ste2p model showed helix-helix interactions that were consistent with biochemical data. For example, Ste2p has a GXXXG motif in H1 that includes Gly56 and Gly60. These motifs mediate helix-helix interactions38 and have been implicated in the dimerization of Ste2p10. To achieve this, these glycines must face outwards the protein. In our model, these glycines are properly oriented (see Figure 3).

Residues involved in helix-helix interactions in Ste2p model. The transmembrane helices of Ste2p are presented as ribbons and are enumerated from H1 to H7. For convenience, transmembrane helices are displayed in a clockwise orientation. Hydrogen bonds are formed between Arg58-Tyr101, Ser170-Glu143, and Ser107-Gln51-Thr110, and are displayed with dots. Gly56 and Gly60 are oriented outwards the protein. The image was generated with PyMol.
As opposed to globular proteins, interactions between transmembrane helices are generally mediated by polar amino acids located in the transmembrane region39. These amino acids can form single helix-helix contacts or hydrogen-bonding networks. It has been proposed based on sequence conservation that the H1-H2 interaction is mediated by a hydrogen bond between Arg58 (HI) and His94 (H2)27. However, in our Ste2p model Arg58 may form a hydrogen bond with Tyr101 on the same face of H2 (see Figure 3), so further mutagenesis and structural experiments may test for the relevance of these pairs of residues in the H1-H2 interaction. Similarly, our model proposed other new helix-helix interactions. For instance, the proximity of residues Ser170 (H3) and Glu143 (H4) would mediate H3-H4 interactions (see Figure 3). Likewise Ser107 and Thr110 of the extracellular loop 2 may interact with Gln51 (H1) (see Figure 3). Our three-dimensional model of Ste2p also displays the orientation of residues that are consistent with their functional role in other GPCRs. For instance, we observed that Gln149 appears to be functionally equivalent to Glu134, which is part of the ERY motif in rhodopsin. Both Glu134 in rhodopsin and Gln149 in Ste2p are localized in a similar position at the end of H3, and are residues where mutation to arginine or alanine leads to constitutive activation40–42. In agreement with these previous observations, Gln149 is oriented in a position similar to that of Glu134 in rhodopsin and presumably shares the role of preventing constitutive activation of the receptor; the reported single point mutations to alanine in positions Gln149 or Asn84 rendered a constitutive activation of the Ste2p receptor, and a double mutation that swapped the side chains of these two residues maintained a wild-type phenotype suggesting these residues are in close proximity in the 3D structure of Ste2p. In our model, Gln149 and Asn84 are 10.5Å apart and facing away from each other, indicating that our model is not consistent with these experimental results. Since our model did not include the N and C termini located at the side where Gln149 lies, it seems that the orientation of H2 and H3 on this side of the transmembrane region of the receptor are not properly oriented in our model. Yet, pheromone binding takes place at the opposite side of the transmembrane region and presumably does not include these helices, so we proceed to analyze the reliability of our model.
Docking. The ligand binding orientations vary widely between the members of the GPCR family43. It has been shown that large ligands bind to the extracellular loops of GPCRs, while small ligands bind to the transmembrane region of receptor. In the case of peptide ligands it has been proposed that a combination of both binding modes takes place. In such case, the ligand binds first to extracellular loops and later enters the transmembrane domain where the binding pocket acts like an epicenter of conformational changes. These conformational changes are propagated to intracellular loops through the movement of helices. Then, the intracellular domain binds to and activates the trimeric G protein43. In the case of Ste2p, alpha pheromone binding has been mapped to the pocket formed by the extracellular ends of the transmembrane helices, specifically H1, H5, and H617,18,33,34.
The GPCRs interact with their ligands via hydrogen bonds, ionic pairs and hydrophobic contacts44. The interactions between Ste2p and the alpha pheromone responsible for binding and activation are not well characterized. However, electrostatic interactions have been suggested to play a critical role in the Ste2p-alpha pheromone complex45.
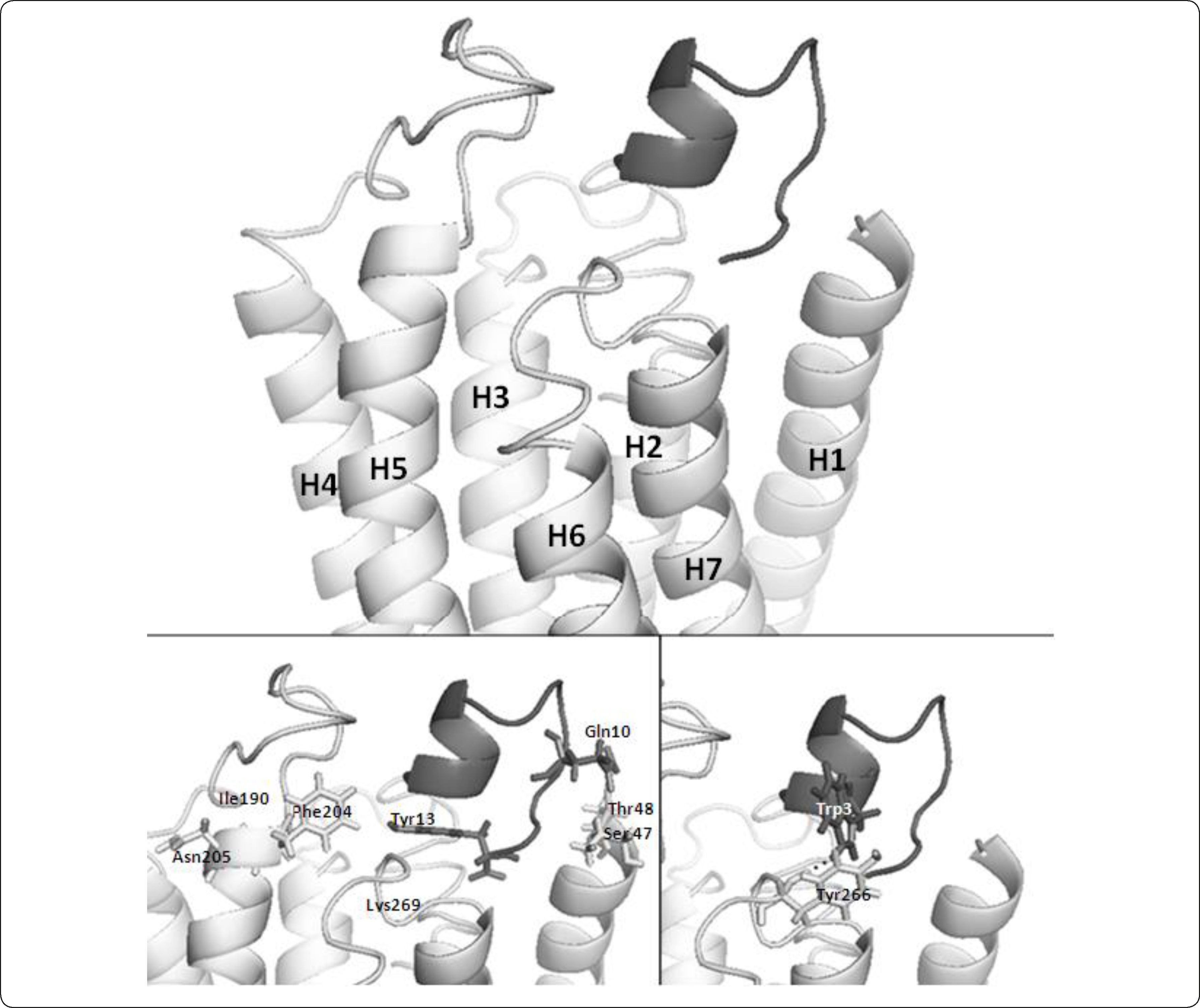
The docking simulation aimed to model the Ste2p-alpha pheromone interaction (Figure 4). In this model all contacts in the Ste2p-alpha pheromone interaction previously reported are present. For instance, previous studies about the affinities and activities of various alpha pheromone analogues have shown that residues Ser47 and Thr48 are near Gln10 in the alpha pheromone17. As we see in Figure 4, our binding model is congruent with these data. Phe204 and Tyr266 are located in the extracellular ends of the helices H5 and H6, respectively. These helices are linked at the opposite end involved in pheromone binding by the third intracellular loop that interacts with the trimeric G protein and promotes its activation14,46,48. Site-directed mutagenesis studies showed that Phe204 interacts with the C-terminal region of the pheromone (likely with Tyr13), while Tyr266 interacts with the N-terminal region (likely with Trp1 or Trp3)18. Both residues, Phe204 and Tyr266, play roles in the transformation of Ste2p into an activated state upon agonist binding18,47. As shown in Figure 4 our model is consistent with these experimental data despite the previously noted differences in our model about H6 and the intracellular ends of H2 and H3 (residues Asn84 and Gln149). Finally, interaction between aromatic residues in the alpha pheromone and Ste2p could be stabilized by ring stacking effects or by hydrogen bonding. In our model the interaction between Tyr266 and Trp3 is stabilized by hydrogen bonding (Figure 4).
 binds to the receptor residues Ser47 and Thr48 (H1)17. Then, the central region of the pheromone (Pro8-Gly9) forms a β-turn46. At this point, Phe204 (H5) is near to Tyr13 in the pheromone18, and Lys269 (H6) is near to both the N and C-termini of the pheromone. Asn205 is facing toward the outside of Ste2p and away from the binding pocket (left). In our model, Tyr266 (H6) is oriented towards the surrounding lipids and is inaccessible to solvent. This orientation is consistent with experimental data from18. Finally, Tyr266 interacts with Trp3 in the pheromone, mediating the transition to an active state of Ste218,47 (right). The image was generated with PyMol.")
Atomic three-dimensional model of the interaction between Ste2p and the alpha pheromone. Ste2p is shown in a light grey ribbon representation. The alpha pheromone is shown as a dark grey object. The upper part of the figure illustrates the binding pocket formed by extracellular loops and extracellular ends of H1, H5, and H6. In the lower part of the figure, selected residues of Ste2p and pheromone are shown in a ball-and-stick representation. First, the C-terminal region of the pheromone (Gln10) binds to the receptor residues Ser47 and Thr48 (H1)17. Then, the central region of the pheromone (Pro8-Gly9) forms a β-turn46. At this point, Phe204 (H5) is near to Tyr13 in the pheromone18, and Lys269 (H6) is near to both the N and C-termini of the pheromone. Asn205 is facing toward the outside of Ste2p and away from the binding pocket (left). In our model, Tyr266 (H6) is oriented towards the surrounding lipids and is inaccessible to solvent. This orientation is consistent with experimental data from18. Finally, Tyr266 interacts with Trp3 in the pheromone, mediating the transition to an active state of Ste218,47 (right). The image was generated with PyMol.
However, our model does not present the Asn205-pheromone contact. Asn205 has been implicated in the binding of the N-terminal domain of the pheromone because mutations at this position affect the Ste2p binding to the pheromone33. It has been shown that residues that are important for binding are not necessarily near the ligand or at the interface in the three-dimensional structure49. In our Ste2p model, Asn205 faces away from the binding pocket (Figure 4). Thus, according to our model, Asn205 would contribute to binding not by direct interaction with the pheromone. In such case, it would be expected that Asn205 may contribute to the pheromone binding through a set of interactions with residues that support the position of the residues that directly bind to the pheromone. Analyzing the contacts (cutoff distance <= 5Å) of the Ste2p residues involved in the pheromone binding (Ser47, Thr48, Phe204 and Tyr266) and comparing these with the contacts of residue Asn205, we learned these share 9 residues (Val191, Tyr193, Asn194, Tyr203, Phe204, Ala206, Ser207, Thr208, and Ile209), which corresponds to 29% of the residues that support the position of the residues directly binding the pheromone. This analysis further supports the relevance of Asn205 in pheromone binding through indirect contacts with the ligand binding residues. These results constitute a hypothesis that can be experimentally tested by performing double swap mutants at Asn205 and the residues in the pheromone proposed to interact with Asn205 for instance; in the present study we decided to mutate another region of the protein as explained below.
Additionally, our three-dimensional model has some discrepancies with other computational studies reported previously. For example, Phe204 does not interact with Tyr13 of the alpha pheromone in the Umanah model34. The lack of interaction between these residues was modeled based on studies of alpha pheromone analogues and chemical cross-linking experiments that proposed an interaction between Tyr13 of the alpha pheromone with Arg58 of Ste2p19. Our model on the other hand, includes the interaction between Tyr13 and Phe204 proposed by Lee and colleagues17, based on site-directed mutagenesis and cross-linking experiments that were disregarded by the group of Umanah and colleagues. We prefer to use the Tyr13-Phe204 interaction in the construction of our Ste2p model because it has more support by other research groups18,50–52 than the Tyr13-Arg58 interaction19,34.
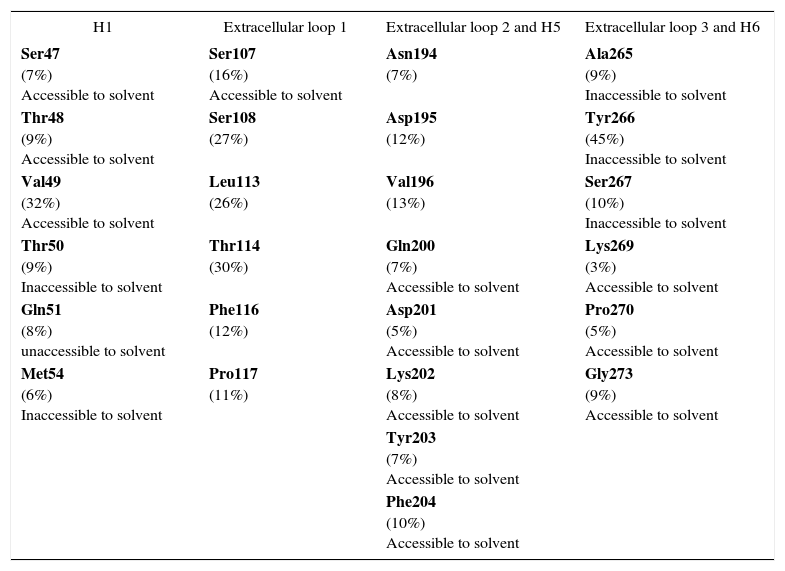
In the interface we found residues both accessible and inaccessible to solvent. This is a feature of many other interfaces of proteins, where solvent accessible residues play roles in ligand binding, while those not accessible to solvent mediate the transition to an active state52. Additionally, our binding model allows us to propose that the interface between Ste2p and the pheromone is composed of 26 residues. As shown in Table I, interface residues include both polar and non-polar residues. This is consistent with previous studies indicating that both hydrophobic and hydrophilic residues form the binding pocket of Ste2p53. The interface residues that are structurally and functionally important tend to be conserved residues or they have a lower rate of mutation compared with the rest of the residues of the protein54. This is the case for Tyr266, which is a critical residue for binding as well as signal transduction18,47 and shows a large percentage of sequence identity conservation (see Table I). However, this is not always the case and many residues that are important for binding are not conserved55. In agreement with this idea, Table I shows that few interface residues are conserved among Ste2p orthologues (Val49, Ser108, Leu113, Thr114, Tyr266). Furthermore, Ser108 and Thr114 of the first extracellular loop, found in the interface of Ste2 and the alpha pheromone in this study, were previously proposed as critical for signaling16. Gln51 and Ser107, also found at the interface, are hydrogen bonded as suggested by the Ste2p model developed in this work, yet these positions do not preserve the same side chain as Ste2p in other fungal orthologues. Considering that the first extracellular loop has been observed to undergo a conformational change upon ligand binding15 this hydrogen bond could be modified during the signaling process. Other non-conserved residues (e.g., Val49, Asp201) were also found in the interface that could play a critical role in Ste2p function. Site-directed mutagenesis on these residues may help to test the role of these residues in ligand binding or signaling.
Composition of the interface between Ste2 and alpha pheromone. Interface residues were defined as those residues in Ste2p that were no more than 5Å apart from the pheromone. Solvent accessibility data were obtained from Lin, J.C., et al., 200453. Percentage of conservation (indicated in parenthesis for each residue; this percentage represents the fraction of 214 orthologue sequences that have an identical residue to Ste2p at that position) was derived from the multiple sequence alignment for all the Ste2p orthologues reported for PFAM family PF02116. Those positions with the largest percentage of sequence identity conservation are marked in bold.
| H1 | Extracellular loop 1 | Extracellular loop 2 and H5 | Extracellular loop 3 and H6 |
|---|---|---|---|
| Ser47 (7%) Accessible to solvent | Ser107 (16%) Accessible to solvent | Asn194 (7%) | Ala265 (9%) Inaccessible to solvent |
| Thr48 (9%) Accessible to solvent | Ser108 (27%) | Asp195 (12%) | Tyr266 (45%) Inaccessible to solvent |
| Val49 (32%) Accessible to solvent | Leu113 (26%) | Val196 (13%) | Ser267 (10%) Inaccessible to solvent |
| Thr50 (9%) Inaccessible to solvent | Thr114 (30%) | Gln200 (7%) Accessible to solvent | Lys269 (3%) Accessible to solvent |
| Gln51 (8%) unaccessible to solvent | Phe116 (12%) | Asp201 (5%) Accessible to solvent | Pro270 (5%) Accessible to solvent |
| Met54 (6%) Inaccessible to solvent | Pro117 (11%) | Lys202 (8%) Accessible to solvent | Gly273 (9%) Accessible to solvent |
| Tyr203 (7%) Accessible to solvent | |||
| Phe204 (10%) Accessible to solvent |
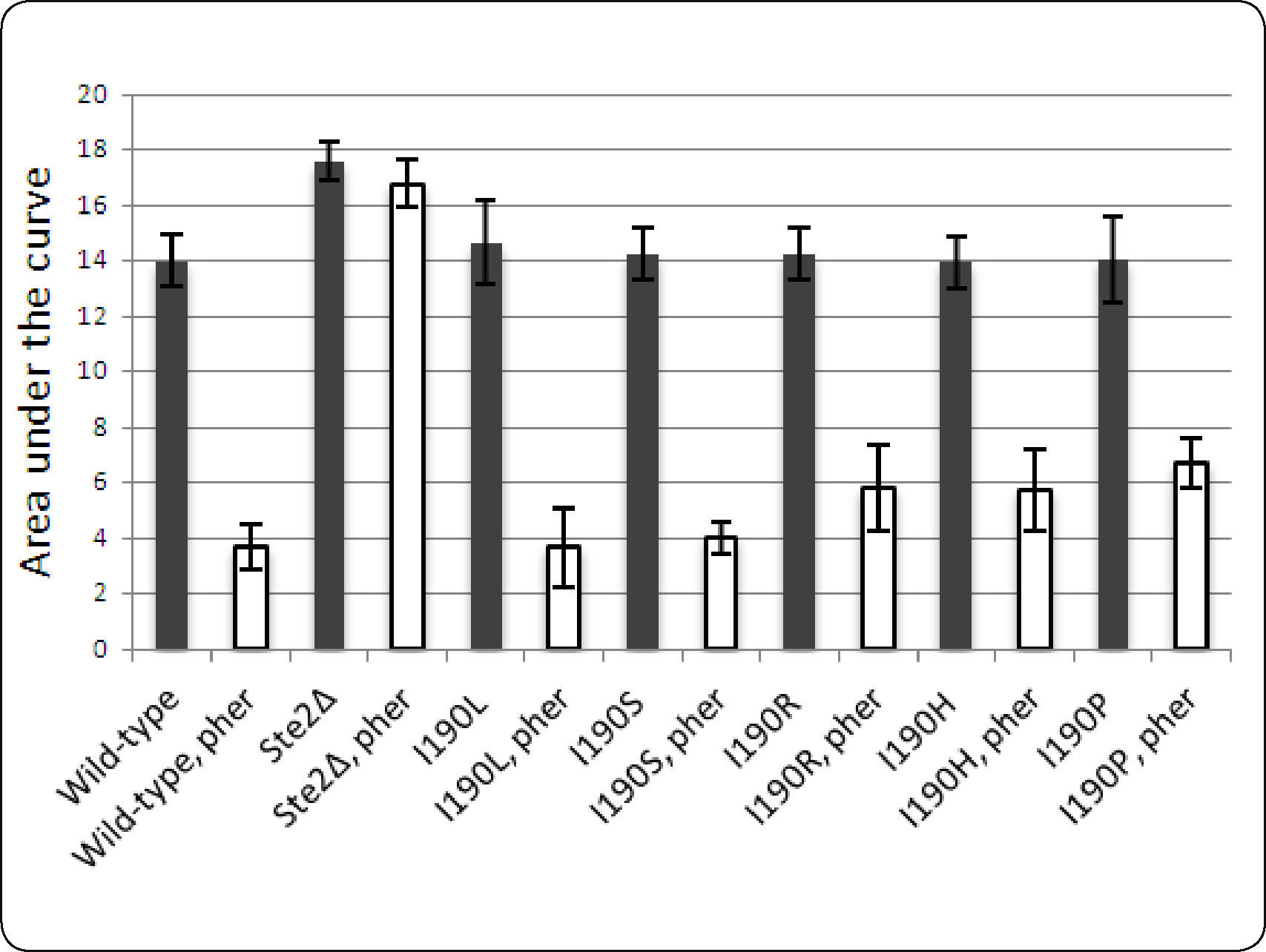
Site-directed mutagenesis of STE2. Several studies have shown that the second extracellular loop of GPCRs plays an important role in ligand binding21–25. Since no mutation for Ste2p at this loop has been reported, we did not use this information to guide the docking of the pheromone to Ste2p. A sequence conservation analysis performed among fungal receptors (http://www.yeastgenome.org/cache/fungi/YFL026W.html) indicated that residue Ile190 of extracellular loop 2 might be relevant for Ste2p activity. In this study, Ile190 was mutated to test for its role in Ste2p function. Five mutants (Ile190Arg, Ile190His, Ile190Pro, Ile190Leu and Ile190Ser) were obtained and their ability to respond to the pheromone was tested. Figure 5 shows the cell growth of all strains expressing these mutants. Two mutations that introduced a ring into that position (Ile190His, Ile190Pro) or arginine reduced slightly the cell growth arrest induced by the pheromone, while two other mutations (Ile190Leu, Ile190Ser) did not reduce the cell growth arrest induced by the pheromone. These results indicate that this position is not critical for receptor function, but ring and positively-charged side chains are tolerated less at this position. In our Ste2p-alpha pheromone model, Ile190 is oriented away from the binding pocket, behind Lys202, a direct ligand of the pheromone (Table I). Placing an arginine at position 190 could result in an excess of positive charge at this site, altering the conformation of Lys202 and debilitating the interaction with the pheromone. Placing another ring, such as Pro or His, at position 190 could modify the position of Tyr203, another residue in direct contact with the pheromone, and/or interact with His126 at the N-terminus of H3 (Figure 1), altering the tilt of H3 with allosteric consequences at the cytoplasmic loops that could impair signaling.
 is represented by the Area Under the growth curve (Y axis) both in the presence and absence of the alpha pheromone (indicated by the abbreviation “pher” in the figure). As control, the growth of wild type strain (BY4741) and a strain carrying a deletion of STE2 gene are presented.")
Area under the growth curve for each mutant strain. The population growth of yeast strains expressing any of five mutants at Ile190 (I190L, I190S, I190R, I190H and I190P) is represented by the Area Under the growth curve (Y axis) both in the presence and absence of the alpha pheromone (indicated by the abbreviation “pher” in the figure). As control, the growth of wild type strain (BY4741) and a strain carrying a deletion of STE2 gene are presented.
Validating a model requires testing four different statistical parameters: true positive (TP), true negative (TN), false positive (FP) and false negative (FN) predictions. Yet, most modeling studies of protein structure commonly test for TP; for instance, the world contest on protein structure prediction only tests for TP by establishing a score that accounts for the fraction of the model that may be superimposed to the real protein structure with no more than certain cutoff RMSD value56. The relevance of the other 3 parameters becomes apparent when it is recognized that experimental protein 3D structures should also be considered a model to test for the critical role of residues in protein function. In the present work we tested 4 parameters in our model, thus providing a more complete test about the quality of the generated structure. Particularly, we showed that:
TP are residues predicted critical and indeed are critical for protein function/structure. For instance, we show that residues Phe204 and Tyr266 that are close to the pheromone in our model are indeed important for binding.
FP are residues predicted critical but are not truly relevant for protein function/structure. Here we show that residue Ile190 is predicted by sequence conservation to be relevant for protein structure/function, yet our experimental results provide evidence against this hypothesis.
TN are residues predicted not critical and are not truly relevant for protein function/structure. Our 3D model predicted that Ile190 was not critical and here we show that indeed this is the case.
FN are residues that are predicted not critical but are truly relevant for protein function/structure. Our model predicts that residues Gln149 and Asn84 are far apart from each other, yet experimental evidence has shown otherwise.
Note that TN and FN are difficult to test in protein structure-function studies because in most cases only residues with an effect on protein structure or function are reported in the literature. Yet, we argue this information is particularly relevant in testing for structure-function prediction models, such as in the case of residues like Ile190.
CONCLUSIONSThe present study presents an atomic three-dimensional structure model of the alpha pheromone receptor from S. cerevisiae, Ste2p, based on state-of-the-art modeling methods and current available experimental data. The Ste2p model is consistent with most available biochemical information and allows us to propose specific interactions that could stabilize the native conformation of the receptor. Furthermore, a model of the interaction between Ste2p and the alpha pheromone was generated based on what is known about the binding contacts. The Ste2p-alpha pheromone model allowed us to identify 26 residues that are presumably found in the interface, but did not include Ile190, a conserved residues among fungi species. Mutagenesis of Ile190 in Ste2p had small or no effect on receptor function and in the model this residue points away from the pheromone. Thus, the model may serve as a starting point for further site-directed mutagenesis to test the structure-function relationship of this receptor.
This work was in part supported by PAPIIT grant number IN208014 to Gabriel del Río. Laura Marina Robles (fellowship number 261023) was supported in part by Consejo Nacional de Ciencia y Tecnología (CONACYT) while she was student of the Programa de Maestría en Ciencias Bioquímicas, UNAM. We thank María Teresa Lara Ortiz at the Institute of Cellular Physiology/Universidad Nacional Autónoma de México for her technical assistance in this work.