









La terapéutica de la esclerosis múltiple ha avanzado notablemente en los últimos años al tiempo que ha aumentado su complejidad. El propósito de este documento de consenso es presentar recomendaciones y pautas concretas sobre la estrategia a seguir en el tratamiento para modificar el curso de la esclerosis múltiple.
Material y métodosexpertos en el tratamiento y en investigación clínica en esclerosis múltiple propuestos por el grupo de enfermedades desmielinizantes de la Sociedad Española de Neurología (SEN) elaboraron un documento inicial con recomendaciones para el tratamiento de esta enfermedad. La versión final de este documento fue remitida a los miembros del grupo de enfermedades desmielinizantes de la SEN, quienes pudieron realizar modificaciones y sugerir cambios al manuscrito final. Tras considerar estas enmiendas, el comité de expertos validó el documento final.
Resultados y conclusionesse revisan los niveles de evidencia y las indicaciones para el tratamiento de las diferentes formas clínicas de esclerosis múltiple y se hacen recomendaciones de uso de los medicamentos. Además de los fármacos autorizados, se revisan también otros productos ocasionalmente empleados, así como la terapia combinada, los criterios de respuesta terapéutica, los niveles de cambio de tratamiento y finalmente se hace una propuesta de escalado terapéutico.
Treatment of multiple sclerosis has advanced considerably in the last few years, at the same time as its complexity has increased. The purpose of this consensus document is to provide specific recommendations and rules on the strategy to follow in the treatment of multiple sclerosis in order to modify its course.
Material and methodsExperts on the treatment and clinical research on multiple sclerosis proposed by the Demyelinating Diseases Group of the Spanish Neurology Society (SEN) prepared an initial document with recommendations for the treatment of this disease. The final version of this document was submitted to members of the Demyelinating Diseases Group of the SEN, who were able to make modifications and suggest changes to the final manuscript.
Results and conclusionsA review has been made of the evidence levels and indications for the treatment of the different clinical forms of multiple sclerosis, and recommendations made for the use of drugs. As well as authorised drugs, a review has also been made of other occasionally used products, as well as combined therapy, therapeutic response criteria, levels of treatment changes, and finally a proposal is made on therapeutic escalation.
El propósito de este documento de consenso es presentar recomendaciones y pautas concretas sobre la estrategia a seguir en el tratamiento de la esclerosis múltiple para modificar el curso de la enfermedad, sin entrar en el tratamiento agudo de los brotes, ni en el sintomático o de apoyo. Los autores son expertos en tratamiento e investigación clínica en esclerosis múltiple propuestos por el grupo de enfermedades desmielinizantes de la Sociedad Española de Neurología.
IntroducciónEn 1993, la Food and Drug Administration (FDA) autorizó el primer interferón beta para el tratamiento de la esclerosis múltiple (EM). Estos hechos iniciaron el despegue de la investigación clínica y, cuando finalizaba esa década, se supo la importancia de iniciar el tratamiento lo antes posible para limitar el daño axonal. De forma progresiva, se han ido incorporando fármacos y pautas terapéuticas hasta configurar el arsenal actual de la EM. La medicina basada en la evidencia (MBE) o el empleo de la información derivada de los mejores estudios para tomar decisiones debe completarse con el criterio experto para la elección de la mejor opción terapéutica en un sujeto y un momento concretos.
El tratamiento individualizado en la práctica diaria debe ajustarse a la forma clínica de la enfermedad y su evolución en cada sujeto. Los estudios clínicos seleccionan a los participantes según un fenotipo o el perfil de la enfermedad y proporcionan el criterio del tratamiento más eficaz, mientras que el clínico conoce la evolución de la enfermedad en el paciente concreto. La evidencia ganada en los estudios clínicos considerados decisivos y concluyentes y su valoración por la autoridad reguladora, junto con la experiencia profesional, son condiciones para alcanzar la excelencia clínica. Aunar experiencia personal y evidencia científica es el objetivo de estas recomendaciones.
Para que estas recomendaciones sean robustas, sencillas y prácticas, se atiende básicamente al nivel A de evidencia, que proviene de los estudios de clase I, comparativos contra placebo y con grupo control en paralelo. No obstante, no siempre es fácil trasladar los conocimientos derivados de una situación experimental controlada a la práctica clínica habitual. En los ensayos clínicos, la variabilidad de los sujetos de experimentación debería ser la mínima posible. En condiciones reales, esta variabilidad se amplía y aparecen supuestos no contemplados en la situación experimental. El hecho de que una determinada intervención se haya demostrado eficaz en condiciones experimentales no implica de forma necesaria su eficacia cuando se aplica a la población general. Por el contrario, no todas las situaciones reales son susceptibles de ser analizadas en situación experimental (p. ej., enfermedades de escasa prevalencia, presentaciones excepcionales de enfermedades prevalentes, comorbilidad, polimedicación, etc.), por lo que no todas las decisiones clínicas pueden estar avaladas por resultados validados previamente en ensayos clínicos.
MetodologíaEn este documento de consenso se emplean los tres niveles de evidencia siguientes:
- 1.
Nivel A: estudios clínicos de clase I aceptados por la autoridad reguladora para la autorización del tratamiento: los estudios principales concluyentes y decisivos. Muy recomendable.
- 2.
Nivel B: estudios de clases I o II que apoyan la utilidad y eficacia de la terapia estudiada, pero que la autoridad reguladora no los ha evaluado o no ha considerado concluyentes. Recomendación favorable.
- 3.
Nivel C: estudios de clase III, con evidencia proveniente de estudios descriptivos no experimentales bien diseñados, como los estudios comparativos, estudios de correlación o estudios de casos y controles. Recomendación favorable pero no concluyente.
- 4.
Nivel U: criterio de los expertos basado en experiencias clínicas o documentos u opiniones de comités de expertos.
En la dirección www.emea.europa.eu, se seleccionaron y recuperaron los informes de evaluación científica (European Public Assessment Report [EPAR] —informe de evaluación científico de la EMEA—) de la Agencia Europea del Medicamento (EMEA, European Medicines Agency) de todos los productos autorizados para el tratamiento de la EM. Se identificaron los estudios que la EMEA aceptó como válidos para apoyar la autorización. En MEDLINE y EMBASE se obtuvieron los estudios de clases I y II que no fueron suficientes para soportar una autorización o modificación del tratamiento, pero que han creado opinión entre los expertos en el manejo terapéutico de la EM. En la dirección www.agemed.es de la Agencia Española del Medicamento se obtuvieron las fichas técnicas de los fármacos autorizados para el tratamiento de la EM.
En una reunión inicial, el grupo de expertos acordó el objetivo del documento de recomendaciones y los niveles de evidencia a emplear. Se asignaron las secciones del documento que cada uno debía elaborar y se establecieron el procedimiento y el calendario correspondientes. Tras la realización de diversas reuniones para revisión y enriquecimiento del manuscrito sobre la base del consenso, el grupo revisó el borrador final y consolidó la versión final del documento de recomendaciones. Dicho documento se remitió a los miembros del grupo de enfermedades desmielinizantes de la Sociedad Española de Neurología, quienes pudieron realizar recomendaciones o modificaciones al manuscrito final. Tras considerar estas enmiendas, el comité de expertos validó el documento final.
Evidencia e indicaciones de los tratamientos aprobados en las diferentes formas clínicasSe denomina síndrome desmielinizante aislado (SDA) al primer episodio de disfunción neurológica. Su identificación es importante porque la mayoría de estos pacientes va a desarrollar una EM con el curso de los años. En torno a un 85-90% de los pacientes con EM comienzan con la forma remitente recurrente (EMRR), de los que una proporción significativa evoluciona a la forma secundariamente progresiva (EMSP) pasados 10-15 años desde el inicio de la enfermedad. El 10-15% restante inicia con la forma progresiva primaria (EMPP), con progresión sostenida de la discapacidad. Un número reducido de pacientes presenta un curso progresivo-recurrente (EMPR). La EMEA, desde una perspectiva terapéutica, engloba bajo el término EM recidivante (EMR) todas las formas clínicas que presenten brotes.
Se ha demostrado que determinados fármacos son capaces de modificar el curso de la EM, disminuyendo el número de brotes y de lesiones en la resonancia magnética (RM). Hay más dudas en cuanto si estos mismos fármacos son capaces de modificar la evolución de la discapacidad residual de una forma clínicamente significativa1–5. Hay argumentos que indican que desde el inicio de la enfermedad se produce un proceso de daño axonal, más o menos relacionado con la inflamación6. Este daño axonal sería el sustrato patológico de la discapacidad residual. Por ello, se ha argumentado que un tratamiento desde el inicio clínico de la EM retrasaría la evolución hacia una discapacidad residual significativa.
Recomendaciones generales
- •
Las personas con EM tienen derecho a todos los fármacos disponibles para su forma de enfermedad.
- •
El neurólogo es el responsable del tratamiento de las personas con EM, y debe tener acceso a todos los fármacos.
- •
Los fármacos autorizados para el tratamiento de la EM que tengan distintas posología, vía o forma de administración no son equivalentes.
La primera línea de tratamiento comprende los inmunomoduladores autorizados para su empleo tras establecer el diagnóstico. Con cualquiera de ellos se puede iniciar el tratamiento y son intercambiables entre sí cuando se considera conveniente un cambio de fármaco.
Síndrome desmielinizante aisladoLos criterios de indicación del uso de fármacos inmunomoduladores en el síndrome desmielinizante aislado pretenden identificar a los pacientes con alto riesgo de desarrollar EM. Esto se define en la ficha técnica de los productos aprobados como “pacientes con un único acontecimiento desmielinizante con un proceso inflamatorio activo, si es lo bastante grave como para justificar el tratamiento con corticosteroides intravenosos, si se han excluido diagnósticos alternativos y si resultan tener un riesgo elevado para el desarrollo de esclerosis múltiple definida clínicamente”. El elevado riesgo se define de forma variable en función de la indicación recogida en las fichas técnicas de los productos7–10.
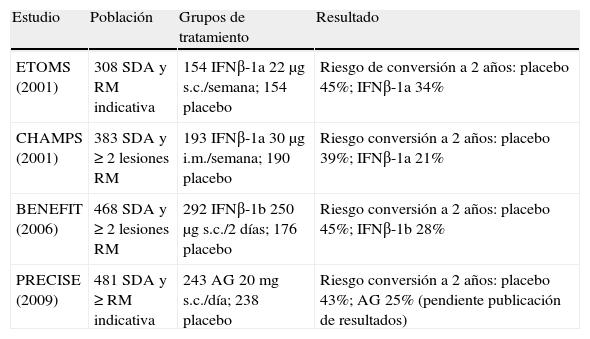
Cuatro estudios han demostrado la eficacia del interferón beta (IFNβ) y el acetato de glatirámero en el tratamiento del SDA (tabla 1). Los tres estudios publicados11–13 reúnen 1.160 casos con SDA y RM indicativa de EM y tratados durante al menos 2 años con IFNβ (n=639) o con placebo (n=521) y con una diferencia en la tasa de conversión a 2 años en torno al 45% en todos ellos. Un metaanálisis14 de los tres estudios ha resultado en una odds ratio (OR) (cociente de probabilidades) conjunta de 0,53, con un intervalo de confianza (IC) del 95% de 0,41-0,71, que evidencia de forma muy significativa (p<0,0001) que el IFNβ retrasa de forma importante la conversión a EM clínicamente definida (EMCD) de los sujetos con SDA. Adicionalmente, estos estudios demostraron que el tratamiento con IFNβ disminuyó el número y el tamaño de las lesiones cerebrales detectadas en la RM. Tras la evaluación de estos resultados, la EMEA y la FDA autorizaron el empleo del IFNβ-1a intramuscular (i.m.) y del IFNβ-1b subcutáneo (s.c.) para el tratamiento del SDA. El estudio de clase I con IFNβ-1a s.c., pese a haber demostrado eficacia, no se presentó en su tiempo a las agencias reguladoras; en el momento de escribir estas líneas se está pendiente de conocer los resultados de otro ensayo con este fármaco en el SDA. Recientemente, la EMEA y la Agencia Española del Medicamento y Productos Sanitarios (AEMPS) han autorizado incluir el acetato de glatirámero en esta indicación (tabla 1 y tabla 2).
Estudios en síndrome desmielinizante aislado (SDA) con nivel A de evidencia.
| Estudio | Población | Grupos de tratamiento | Resultado |
| ETOMS (2001) | 308 SDA y RM indicativa | 154 IFNβ-1a 22μg s.c./semana; 154 placebo | Riesgo de conversión a 2 años: placebo 45%; IFNβ-1a 34% |
| CHAMPS (2001) | 383 SDA y ≥ 2 lesiones RM | 193 IFNβ-1a 30μg i.m./semana; 190 placebo | Riesgo conversión a 2 años: placebo 39%; IFNβ-1a 21% |
| BENEFIT (2006) | 468 SDA y ≥ 2 lesiones RM | 292 IFNβ-1b 250μg s.c./2 días; 176 placebo | Riesgo conversión a 2 años: placebo 45%; IFNβ-1b 28% |
| PRECISE (2009) | 481 SDA y ≥ RM indicativa | 243 AG 20mg s.c./día; 238 placebo | Riesgo conversión a 2 años: placebo 43%; AG 25% (pendiente publicación de resultados) |
AG: acetato de glatirámero; IFN: interferón; i.m.: intramuscular; RM: resonancia magnética; s.c.: subcutáneo.
Recomendaciones en síndrome desmielinizante aislado.
| • Los IFNβ 1a y 1b y el acetato de glatirámero utilizados en el SDA han demostrado mejorar significativamente el riesgo de desarrollar eslerosis múltiple (EM) |
| • La finalidad del tratamiento del SDA es prevenir la conversión a EM y con ello evitar el acúmulo de discapacidad |
| • La presencia de un SDA no implica por sí mismo la necesidad de tratamiento con fármacos modificadores de la enfermedad (FME) |
| • El primer episodio desmielinizante podría ser tratado con FME en los casos con elevado riesgo de conversión a EM |
| • Presentar 3 de 4 criterios de diseminación (DIS) en espacio (criterios de Barkhof) |
| • Criterios DIS en espacio según los criterios de Swanton |
| • Lesiones captantes y no captantes de gadolinio |
| • Síntesis intratecal de IgG, IgM |
| • Cuantos más factores haya, mayor es la probabilidad de conversión y, por lo tanto, mayor también la recomendación de tratamiento |
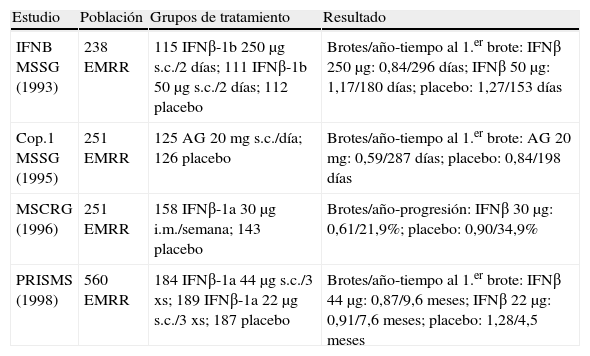
Como la gran mayoría de los sujetos con esclerosis múltiple comienzan con la forma EMRR, la investigación clínica es predominante en esta forma de la enfermedad. De hecho, fue en EMRR en que por primera vez demostraron su eficacia los fármacos modificadores de la enfermedad (FME) y fue la primera de las indicaciones autorizadas. Los sujetos participantes en estos estudios presentaban una enfermedad activa, con al menos dos brotes en los 2-3 años previos, y la eficacia de estos fármacos se sustanció principalmente en una reducción de la frecuencia de los brotes, junto con una disminución de las lesiones cerebrales detectadas mediante RM.
Los fármacos autorizados para el tratamiento de las formas recurrentes de EM son: IFNβ-1b 250μg subcutáneos en días alternos, IFNβ-1a 22 o 44μg subcutáneos 3 veces por semana, interferón (IFN) beta-1a 30μg i.m. una vez por semana, y acetato de glatirámero 20 mg s.c. todos los días.
Los criterios de indicación de tratamiento en las formas EMRR requieren que se haya presentado 2 o más brotes en los últimos 2 años en el caso del IFNβ-1b y acetato de glatirámero, y 2 o más brotes en los últimos 3 años para el IFNβ-1a i.m. El IFNβ-1a s.c. está autorizado para las formas de EM en brotes, y se permite su uso desde el primer brote con apoyo de RM (criterios de McDonald)15,16(tabla 3 y tabla 4).
Estudios en esclerosis múltiple remitente recurrente (EMRR) con nivel de evidencia A.
| Estudio | Población | Grupos de tratamiento | Resultado |
| IFNB MSSG (1993) | 238 EMRR | 115 IFNβ-1b 250μg s.c./2 días; 111 IFNβ-1b 50μg s.c./2 días; 112 placebo | Brotes/año-tiempo al 1.er brote: IFNβ 250μg: 0,84/296 días; IFNβ 50μg: 1,17/180 días; placebo: 1,27/153 días |
| Cop.1 MSSG (1995) | 251 EMRR | 125 AG 20mg s.c./día; 126 placebo | Brotes/año-tiempo al 1.er brote: AG 20 mg: 0,59/287 días; placebo: 0,84/198 días |
| MSCRG (1996) | 251 EMRR | 158 IFNβ-1a 30μg i.m./semana; 143 placebo | Brotes/año-progresión: IFNβ 30μg: 0,61/21,9%; placebo: 0,90/34,9% |
| PRISMS (1998) | 560 EMRR | 184 IFNβ-1a 44μg s.c./3 xs; 189 IFNβ-1a 22μg s.c./3 xs; 187 placebo | Brotes/año-tiempo al 1.er brote: IFNβ 44μg: 0,87/9,6 meses; IFNβ 22μg: 0,91/7,6 meses; placebo: 1,28/4,5 meses |
AG: acetato de glatirámero; IFN: interferón; i.m.: intramuscular; s.c.: subcutáneo.
Recomendaciones en la esclerosis múltiple remitente recurrente (EMRR).
| • La finalidad del tratamiento es prevenir la recurrencia de brotes y el acúmulo de discapacidad |
| • Se recomienda el tratamiento temprano de la esclerosis múltiple (EM), mediante la aplicación de los criterios de McDonald actualizados para el diagnóstico precoz |
| • Como tratamiento de primera línea de la EMRR se recomienda utilizar: IFNβ-1b subcutáneo (s.c.), IFNβ-1a s.c., IFNβ-1a intramuscular y acetato de glatirámero |
| • En España se permite el uso de azatioprina como fármaco de primera línea |
| • Asímismo, el uso de natalizumab está autorizado como fármaco de primera elección en casos de evolución rápida y agresiva |
| • Como tratamiento de segunda línea de la EMRR están indicados natalizumab y mitoxantrona |
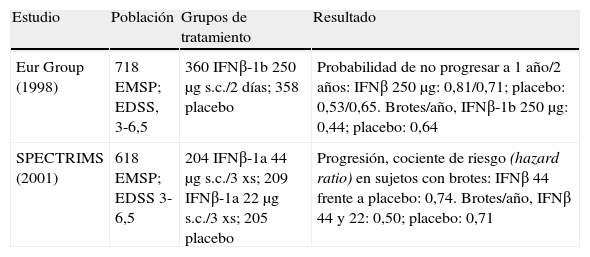
En la actualidad los dos FME autorizados para el tratamiento de la EMSP por disponer de estudios clínicos17,18 que soportan evidencia de nivel A son IFNβ-1b e IFNβ-1a s.c. Las características que deben reunir los sujetos para poder acceder al tratamiento son tener una forma activa de EMSP, definida por8,16:
- •
Haber empeorado en los 2 años previos, de forma progresiva: un punto o más en pacientes con EDSS (Expanded Disability Status Scale) previo < 5,5, o 0,5 puntos en pacientes con EDSS previo de 5,5-6,5.
- •
Haber presentado al menos un brote en los 2 años previos.
En pacientes sin evidencia clínica de brotes, pero en cuya RM se objetive actividad inflamatoria, se podría valorar la indicación de forma individualizada (tabla 5 y tabla 6).
Estudios de clase I en esclerosis múltiple secundariamente progresiva (EMSP).
| Estudio | Población | Grupos de tratamiento | Resultado |
| Eur Group (1998) | 718 EMSP; EDSS, 3-6,5 | 360 IFNβ-1b 250μg s.c./2 días; 358 placebo | Probabilidad de no progresar a 1 año/2 años: IFNβ 250μg: 0,81/0,71; placebo: 0,53/0,65. Brotes/año, IFNβ-1b 250μg: 0,44; placebo: 0,64 |
| SPECTRIMS (2001) | 618 EMSP; EDSS 3-6,5 | 204 IFNβ-1a 44μg s.c./3 xs; 209 IFNβ-1a 22μg s.c./3 xs; 205 placebo | Progresión, cociente de riesgo (hazard ratio) en sujetos con brotes: IFNβ 44 frente a placebo: 0,74. Brotes/año, IFNβ 44 y 22: 0,50; placebo: 0,71 |
IFN: interferón; s.c.: subcutándeo.
Recomendaciones en la esclerosis múltiple secundariamente progresiva (EMSP).
| • La EMSP es susceptible de tratamiento si presenta brotes o actividad inflamatoria en la resonancia magnética |
| • Como tratamiento de primera línea de la EMSP se recomienda la utilización de: IFNβ-1b subcutáneo, IFNβ-1a subcutáneo |
| • Como tratamiento de segunda línea de la EMSP con brotes está aprobado el empleo de mitoxantrona |
| • Podría considerarse el uso de azatioprina en la EMSP con brotes. |
Hasta el momento actual, ningún estudio clínico ha demostrado eficacia en sujetos con EMPP que presentan un deterioro neurológico creciente sin brotes y que suele acompañarse de escasas lesiones en la RM del sistema nervioso central. Por carecer de tratamiento autorizado y por la gravedad del diagnóstico, es el mejor criterio clínico lo que decide el tratamiento en cada caso, tras valorar la razón riesgo/beneficio (tabla 7 y tabla 8).
Fármacos de primera línea e indicaciones autorizadas.
| Fármaco | Posología | Vía | Indicaciones |
| IFNβ-1b (Betaferon®, Extavia®) | 250μg a días alternos | s.c. | SDA que justifica el tratamiento con corticoides inyectados y se considera riesgo alto de desarrollar EM. EMRR con 2 o más brotes en los 2 últimos años. EMSP con brotes |
| IFNβ-1a (Avonex®) | 30μg una vez a la semana | i.m. | SDA que requiere tratamiento con corticoides i.m. y se considera de riesgo alto de desarrollar EM. EMRR |
| IFNβ-1a (Rebif®) | 22 o 44μg 3 días a la semana | s.c. | EMRR según criterios de McDonald. EMSP con brotes |
| Acetato de glatirámero (Copaxone®) | 20mg diarios | s.c. | SDA considerado como riesgo alto de desarrollar EMCD. EMRR con 2 o más brotes en los últimos 2 años y con capacidad de andar sin ayuda |
| Azatioprina (Imurel®) | 2,5 mg/kg/día | Oral | EMRR. Opcional en la EMSP con brotes |
EM: esclerosis múltipe; EMCD: EM clínicamente definida; EMRR: EM remitente recurrente; EMSP: EM secundariamente progresiva; IFN: interferón; i.m.: intramuscular; s.c.: subcutánea; SDA: síndrome desmielinizante aislado.
En EM, un fármaco es de segunda línea cuando su empleo está supeditado al fracaso o la intolerancia de los tratamientos previos de primera línea.
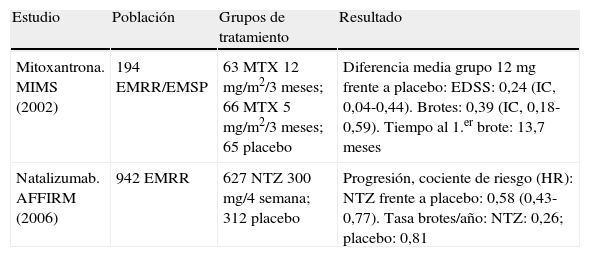
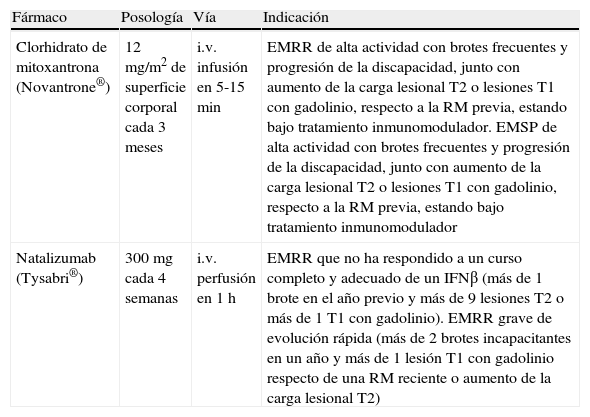
MitoxantronaLa demostración de eficacia frente a placebo de la mitoxantrona (Novantrone®)19 hizo que fuese el primer fármaco autorizado como tratamiento de segunda línea de la EMRR cuando no hay respuesta al tratamiento con inmunomoduladores y persisten la frecuencia de los brotes y las lesiones cerebrales activas evidenciadas en la RM. La mitoxantrona es cardiotóxica, debe administrarse con una fracción de eyección ventricular izquierda > 50% y su empleo requiere control ecográfico o isotópico de la función ventricular izquierda antes y durante el tratamiento. El otro riesgo importante de la mitoxantrona es el desarrollo de una leucemia aguda, por lo que deben realizarse controles hematológicos durante el tratamiento y durante varios años después de finalizarlo. De forma preventiva, la dosis total acumulada no debe superar 140mg/m2. Incluso se recomienda superar la dosis total acumulada de 100mg/m2 sólo en los sujetos que responden y aún presentan signos de actividad20.
NatalizumabEl natalizumab (Tysabri®) es el único anticuerpo monoclonal autorizado para el tratamiento de la EM. Actúa por bloqueo de la integrina α4 leucocitaria y así limita la migración de linfocitos y monocitos a través de la barrera hematoencefálica hacia el sistema nervioso central. Se realizaron dos amplios estudios de fase III en EMRR, uno con natalizumab en monoterapia21 y el otro en tratamiento combinado con IFNβ-1a i.m. en sujetos con EMRR no respondedores al IFNβ-1a i.m., que fue interrumpido por motivos de seguridad por la aparición de casos aislados de leucoencefalopatía multifocal progresiva. Debido a esta circunstancia la EMEA concedió la aprobación de natalizumab como tratamiento para pacientes sin respuesta a fármacos de primera línea o como primera opción en formas de EMRR grave de evolución rápida.
La tabla 9 resume los dos estudios que soportan la autorización de estos dos productos como tratamiento de segunda línea de la EM.
Estudios de clase I con fármacos de segunda línea.
| Estudio | Población | Grupos de tratamiento | Resultado |
| Mitoxantrona. MIMS (2002) | 194 EMRR/EMSP | 63 MTX 12 mg/m2/3 meses; 66 MTX 5 mg/m2/3 meses; 65 placebo | Diferencia media grupo 12mg frente a placebo: EDSS: 0,24 (IC, 0,04-0,44). Brotes: 0,39 (IC, 0,18-0,59). Tiempo al 1.er brote: 13,7 meses |
| Natalizumab. AFFIRM (2006) | 942 EMRR | 627 NTZ 300 mg/4 semana; 312 placebo | Progresión, cociente de riesgo (HR): NTZ frente a placebo: 0,58 (0,43-0,77). Tasa brotes/año: NTZ: 0,26; placebo: 0,81 |
EMRR: esclerosis múltiple remitente recurrente; EMSP: esclerosis múltiple secundariamente progresiva; HR: hazard ratio; IC: intervalo de confianza; MTX: metotrexato; NTZ: natalizumab.
Mitoxantrona y natalizumab son dos productos de eficacia destacable en reducir la progresión de la discapacidad en sujetos con una puntuación en la EDSS de hasta 6 puntos en el límite de capacidad ambulatoria (EDSS < 6), la frecuencia de los brotes y las lesiones cerebrales detectadas en RM. Su perfil de seguridad los ha posicionado como segunda opción, por el riesgo que conllevan y las pruebas y controles que deben instaurarse para minimizarlos.
Debido al distinto perfil de riesgo/beneficio de estos dos fármacos, en la actualidad se considera natalizumab como primera opción en la segunda línea de tratamiento. Ambos fármacos están autorizados como monoterapia y su empleo combinado con los FME de primera línea comporta riesgos adicionales y no cuenta con autorización (tabla 10).
Fármacos de segunda línea y sus indicaciones autorizadas.
| Fármaco | Posología | Vía | Indicación |
| Clorhidrato de mitoxantrona (Novantrone®) | 12 mg/m2 de superficie corporal cada 3 meses | i.v. infusión en 5-15 min | EMRR de alta actividad con brotes frecuentes y progresión de la discapacidad, junto con aumento de la carga lesional T2 o lesiones T1 con gadolinio, respecto a la RM previa, estando bajo tratamiento inmunomodulador. EMSP de alta actividad con brotes frecuentes y progresión de la discapacidad, junto con aumento de la carga lesional T2 o lesiones T1 con gadolinio, respecto a la RM previa, estando bajo tratamiento inmunomodulador |
| Natalizumab (Tysabri®) | 300mg cada 4 semanas | i.v. perfusión en 1 h | EMRR que no ha respondido a un curso completo y adecuado de un IFNβ (más de 1 brote en el año previo y más de 9 lesiones T2 o más de 1 T1 con gadolinio). EMRR grave de evolución rápida (más de 2 brotes incapacitantes en un año y más de 1 lesión T1 con gadolinio respecto de una RM reciente o aumento de la carga lesional T2) |
EMRR: esclerosis múltiple remitente recurrente; EMSP: esclerosis múltiple secundariamente progresiva; IFN: interferón; i.v.: intravenoso; RM: resonancia magnética.
Los fármacos mencionados a continuación no tienen indicación aprobada para el tratamiento de la EM, a excepción de azatioprina. En ellos, los niveles de evidencia son menores o no se han estudio suficientemente.
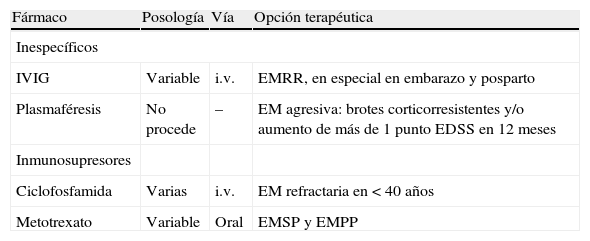
Inmunoglobulinas por vía intravenosaLos estudios clínicos indican que el beneficio principal de las inmunoglobulinas por vía intravenosa (IVIG) es la reducción en el número de brotes, pero la heterogeneidad de los estudios lastra el nivel de evidencia. Un estudio22 amplio con distintas pautas posológicas no ha mostrado eficacia en la reducción de brotes, EDSS o variables de RM. En este contexto, la European Federation of Neurological Societies23 considera las IVIG como tratamiento de segunda línea para la EMRR en caso de intolerancia o enfermedad concomitante. Desestima su uso en SDA y en EMSP. Por último, el tratamiento con IVIG parece ser seguro y probablemente efectivo en la prevención de los ataques durante el embarazo y tras el parto24. Ninguno de los preparados de IVIG está autorizado para tratamiento de la EM, pero pueden considerarse como alternativa en el tratamiento de la EMRR, cuando las terapias convencionales han fracasado, y en los brotes durante el embarazo y en el periodo del posparto.
Azatioprina(Nota: autorizada en primera línea en España, se incluye aquí por ausencia de estudios clase I.)
La azatioprina es un inmunosupresor análogo de la purina que se emplea en la EM desde hace más de 25 años, aunque los estudios son pequeños y con limitaciones metodológicas. Disminuye principalmente los brotes y en menor medida la progresión de la discapacidad25. Cinco estudios agrupados en un metaanálisis26, con datos de 1 a 3 años en que comparaban azatioprina con placebo, muestran que la administración de azatioprina redujo en un 20% el riesgo relativo de que un sujeto presente un brote. También demostró efecto en la disminución de la discapacidad, pero el número de sujetos con ese dato fue escaso. El perfil de seguridad de la azatioprina resulta bueno y el riesgo de cáncer parece relacionado con una duración del tratamiento por encima de 10 años y una dosis acumulada superior a 600 g. Considerando la razón riesgo/beneficio, azatioprina podría ser una alternativa terapéutica, pero falta la confirmación en un estudio clínico con nivel A de evidencia.
CiclofosfamidaCiclofosfamida es un antineoplásico potente que se ha empleado en la EM resistente al tratamiento. Los esquemas terapéuticos ensayados en EM van desde dosis altas intravenosas de inducción con ciclofosfamida y ACTH en 2-3 semanas y seguimiento hasta 2 años hasta dosis bajas de recuerdo cada 2 meses durante 2 años de forma ambulatoria27,28. La pauta de dosis bajas de recuerdo durante 2 años alargó de forma significativa el tiempo hasta la progresión sostenida de la discapacidad en los sujetos menores de 40 años, pero no en los mayores de esa edad. En conjunto, los estudios han demostrado que la administración ambulatoria de ciclofosfamida es segura en sujetos con EM, sin que se objetivara un beneficio clínico importante.
MetotrexatoMetotrexato es un agente antineoplásico inhibidor de la dihidrofolato reductasa, que tiene la ventaja de su administración oral y el inconveniente de su toxicidad hepática. Una revisión reciente29 de los estudios clínicos con metotrexato en EM identificó dos estudios aleatorizados y controlados. Sólo uno de éstos tenía robustez metodológica y seguimiento superior a 3 meses. Este estudio aleatorizado y controlado con placebo en 60 sujetos con EMSP y EMPP detectó que el tratamiento con metotrexato sólo produjo efectos secundarios menores, disminuyó la progresión de la discapacidad y redujo el número de brotes, pero sin alcanzar significación estadística, tras 36 meses de seguimiento. No hay estudios con metotrexato en la EMRR.
PlasmaféresisLa plasmaféresis es una opción terapéutica para determinados pacientes con una EM agresiva que no responde al tratamiento, y pretende eliminar del plasma los agentes proinflamatorios causantes de la agresividad de la EM. Los datos publicados son pocos y se trata de casos individuales con brotes graves resistentes al tratamiento con corticoides o de formas progresivas con aumento rápido de la discapacidad a pesar del tratamiento30. Es una opción terapéutica de evidencia limitada y que puede considerarse en casos individuales de brotes catastróficos y en casos de EM agresivas, con progreso rápido de la discapacidad, a pesar del tratamiento. Recientemente se ha utilizado como parte del tratamiento de la leucoencefalopatía multifocal progresiva asociada a natalizumab.
Nuevas terapias en desarrolloHay varios productos en fase avanzada de desarrollo con estudios principales de clase I, para obtener evidencia de nivel A, que la autoridad reguladora pueda considerar suficiente para autorizar su empleo en el tratamiento de la EM. La gama de nuevos productos incluye desde anticuerpos monoclonales (alemtuzumab, rituximab o daclizumab) hasta nuevas formas de productos ya estudiados en EM, como la cladribina oral, o laquinimod, pasando por inmunosupresores inhibidores de la síntesis de pirimidina (teriflunomida) y agonistas de la esfingosina 1P (fingolimod). Sin adelantar valoraciones de la eficacia, sí que va a ser importante contar con nuevos productos para administración oral (tabla 11).
Fármacos no aprobados y su opción terapéutica.
| Fármaco | Posología | Vía | Opción terapéutica |
| Inespecíficos | |||
| IVIG | Variable | i.v. | EMRR, en especial en embarazo y posparto |
| Plasmaféresis | No procede | – | EM agresiva: brotes corticorresistentes y/o aumento de más de 1 punto EDSS en 12 meses |
| Inmunosupresores | |||
| Ciclofosfamida | Varias | i.v. | EM refractaria en < 40 años |
| Metotrexato | Variable | Oral | EMSP y EMPP |
EM: esclerosis múltiple; EMPP: EM progresiva primaria; EMRR: EM remitente recurrente; EMSP: EM secundariamente progresiva; i.v.: intravenoso.
La terapia combinada pretende cubrir de forma concurrente más de un mecanismo fisiopatológico de la EM para aumentar la eficacia. Sin embargo, no hay ninguna combinación terapéutica autorizada ni estudios clínicos que proporcionen nivel de evidencia A.
La decisión de combinar tratamientos la toma el médico ante un caso concreto, según su experiencia y mejor criterio. La otra situación es la búsqueda de la mejor combinación terapéutica en grupos de pacientes dentro de estudios clínicos con el diseño necesario para obtener resultados decisivos y concluyentes. Una revisión reciente31 de las combinaciones terapéuticas en EM ha identificado al menos 95 combinaciones diferentes e indica expresamente que la calidad de los estudios es limitada. La interrupción por razones de seguridad de dos estudios de clase I con natalizumab combinado con IFNβ-1a i.m. (estudio AFFIRM) y con acetato de glatirámero (estudio GLANCE) ha frustrado las primeras oportunidades de obtener nivel A de evidencia de dos combinaciones terapéuticas para la EMRR. Asimismo, ha reforzado la importancia de la seguridad de las combinaciones terapéuticas. Un balance muy cuidadoso entre eficacia y seguridad y una información muy detallada a los pacientes deben dirigir las decisiones de los clínicos en este tema32,33.
Criterios de respuesta terapéuticaEl curso irregular e impredecible de la EM y la ausencia de tratamientos curativos dificultan concretar criterios de respuesta y fracaso del tratamiento. El perfil basal de la EM o las características del sujeto no permiten avanzar la respuesta33. En principio, es más fácil detectar el fracaso. La respuesta incluye el efecto placebo, el fenómeno de regresión a la media o la evolución natural de la enfermedad y la adherencia al tratamiento, además de los brotes y la evolución de la discapacidad. Los tratamientos autorizados han demostrado que mejoran la evolución de la enfermedad en los primeros años de su administración y queda pendiente conocer su efecto a largo plazo. No obstante y por razones éticas, de eficiencia y seguridad, el manejo clínico de los pacientes con EM obliga a emplear criterios de respuesta o fracaso para adoptar decisiones terapéuticas en un momento y un sujeto concretos.
Para poder valorar su eficacia se considera necesario realizar un curso completo de tratamiento durante un plazo estimado de 6 a 12 meses.
Monitorización de la respuestaPara identificar la respuesta disponemos de parámetros clínicos (brotes y discapacidad) y de RM. Estas variables tienen distinto peso según la forma de la enfermedad, lo que es importante para la actitud terapéutica.
BrotesLos brotes son un buen marcador clínico de actividad, a pesar de los factores de confusión como la regresión a la media o la dificultad para calibrar su gravedad y que su relación con la discapacidad a medio o largo plazo no es lineal34,35. Finalmente, la tasa de brotes está muy influida por cómo se realiza el seguimiento de los pacientes. Si el seguimiento es más frecuente, hay mayor posibilidad de detectar los brotes. Por lo tanto, aunque los brotes nos ofrecen una medida de actividad de la enfermedad, su utilización como medida única de respuesta al tratamiento inmunomodulador debe valorarse con cautela.
Progresión de la discapacidadLa RM muestra que hay más actividad inflamatoria cerebral de la que refleja la clínica de la EMRR36. Esta actividad inflamatoria silente puede producir una progresión del déficit neurológico detectable en exploraciones sucesivas como marcador de una mala respuesta terapéutica. Se considera que el incremento de un punto en la EDSS es un cambio significativo en el estado neurológico, sobre todo si es por encima de 3 de la EDSS. Los incrementos deben confirmarse en el tiempo para desestimar incrementos falsos o transitorios debidos a la recuperación incompleta tras un brote de la enfermedad o bien a otros procesos médicos como fiebre, etc. Un estudio objetivó que el incremento de 1 punto en la EDSS, confirmado a los 6 meses, ofrecía una sensibilidad del 77% y una especificidad del 89% para detectar un incremento de discapacidad a largo plazo37. La EDSS está universalmente consolidada y facilita la comparación entre resultados. Debe considerarse el patrón de valoración y seguir empleándose, con independencia de que se utilicen otras escalas.
Lesiones cerebralesLa RM cerebral detecta zonas hiperdensas, cuya aparición guarda relación con la actividad clínica, tanto en los brotes como en la discapacidad. La asociación entre la RM y las variables clínicas no es muy estrecha y la RM aún no se acepta para valoración subrogada de la eficacia en los estudios para nivel A de evidencia. Sin embargo, los cambios objetivados en la RM son útiles para valorar el efecto de un tratamiento.
Definición de respuestaLas variables clínicas, brotes y discapacidad, determinan la respuesta al tratamiento. La RM es una prueba complementaria de apoyo para la toma de decisiones terapéuticas.
Variables clínicasDisponer de una definición de respuesta validada es fundamental para poder identificar a los sujetos que continuarán con actividad tras una falta de respuesta inicial al tratamiento y que pueden beneficiarse de un cambio terapéutico precoz, cuando todavía la enfermedad no ha causado un daño irreversible. Pero no hay una definición validada de respuesta. Por eso la mejor opción disponible es emplear los criterios de los estudios clínicos que la autoridad reguladora ha aceptado como evidencia suficiente. Esto implica valorar la respuesta ajustada al intervalo de 2 años de los estudios clínicos, que parece corto para una enfermedad crónica, pero es la mejor aproximación.
En cuanto a los brotes, se considera que los sujetos tienen buena respuesta cuando presentan menos de 1 brote en 2 años de tratamiento, que corresponde a una tasa anualizada de 0,5, que es la mitad del criterio de inclusión en los estudios. El tiempo hasta el siguiente brote no se considera un buen parámetro.
En cuanto a la discapacidad, se considera fracaso del tratamiento el aumento de 1 punto en la EDSS, de forma mantenida durante 6 meses consecutivos, en los sujetos con una EDSS ≤ 5,5. Cuando la EDSS es > 5,5 se considera fracaso el aumento de 0,5 puntos. El tiempo transcurrido hasta el empeoramiento sostenido también es un parámetro aceptable para valorar la respuesta. La proporción de sujetos con progresión en un determinado tiempo no es aplicable a un caso concreto en la práctica diaria. Se considera que la respuesta es aceptable si no se aprecia incremento de discapacidad en las magnitudes señaladas.
Variables radiológicasLa RM también informa sobre la actividad de la enfermedad, pero la autoridad reguladora no la considera una variable subrogada. Sin embargo, presentar una lesión T1 (tiempo 1 de relajación) hiperintensa tras gadolinio y/o más de dos lesiones T2 (tiempo 2 de relajación) nuevas respecto de la RM realizada 1-2 años antes indica mala respuesta al tratamiento. La RM puede ser el criterio que determine la opción terapéutica cuando la clínica no es concluyente.
Variables combinadasAunque parece razonable definir la respuesta con una combinación de brotes y discapacidad, se carece de experiencia y no es fácil. Una combinación clínico-radiológica añade el inconveniente de que la RM no se acepta como parámetro subrogado a la clínica. Cada paciente es un caso independiente y el neurólogo considerará cómo priorizar las variables de respuesta en un caso y un momento concretos para decidir la oportunidad de cambiar de tratamiento.
En conclusión, el objetivo principal es prevenir la discapacidad en el largo plazo, por eso, aunque el nivel A de evidencia prima la frecuencia de los brotes, debe ponderarse bien la acumulación de la discapacidad y apoyarse en las lesiones cerebrales de RM y la atrofia cerebral, siempre que se pueda. La evolución de la actividad de la EM es el criterio de ajuste individualizado del tratamiento.
Cambio de tratamiento en la esclerosis múltipleEn la actualidad los medicamentos para tratar la EM pueden clasificarse en cuatro grupos:
- 1.
Medicamentos aprobados de primera línea: IFNβ-1b, IFNβ-1a, acetato de glatirámero (AG) y, en España, azatioprina.
- 2.
Medicamentos aprobados de segunda línea: mitoxantrona y natalizumab.
- 3.
Medicamentos no aprobados, pero sobre los que hay experiencia clínica: ciclofosfamida, metotrexato, inmunoglobulinas i.v., esteroides y asociaciones de medicamentos.
- 4.
Medicamentos en investigación registrados para otras indicaciones: cladribina, micofenolato de mofetilo, rituximab, alemtuzumab, etc.
Los medicamentos aprobados de primera línea tienen en general un perfil de tolerabilidad razonable, con muy escasa inducción de efectos adversos graves, aunque con frecuentes efectos secundarios leves. Su eficacia ronda el 30% de reducción de la tasa de brotes y en ese porcentaje se agrupan los pacientes con respuesta aparentemente completa, no respondedores y respondedores parciales. Es el grupo con el perfil de seguridad y eficacia más consolidado.
Esta gama de fármacos para la EM permite reemplazar un tratamiento mal tolerado o ineficaz por otro de los disponibles y lograr la mejor relación riesgo/beneficio u optimización del tratamiento. La valoración del riesgo del tratamiento es más fácil a medida que se gana experiencia en la exposición a los fármacos. Lo problemático es valorar el beneficio a falta de tratamiento curativo y con la variabilidad de respuesta inherente a la propia enfermedad y a cada individuo. Éste es el objeto de este consenso y, en la sección anterior, ha quedado definido y cuantificado el límite mínimo de respuesta en cada variable de valoración.
Niveles de cambio de tratamientoLas opciones terapéuticas están clasificadas en los cuatro niveles arriba indicados, que jerarquizan los grupos y el paso al nivel superior es un escalado terapéutico. Por otro lado, cada grupo contiene varios fármacos y preparados con distintas posologías canjeables. Esto resulta en varias decenas de combinaciones terapéuticas a disposición del manejo diario de los sujetos con EM.
Las posibilidades de cambio entre los fármacos de primera línea son las siguientes:
- 1.
Fármacos del mismo tipo: IFNβ.
- 2.
Inmunomoduladores: IFNβ y AG.
- 3.
3 IFNβ, AG y azatioprina.
En el primer grupo, las razones de cambio por intolerancia en relación con la vía de administración son claras y no suelen plantear dudas. Un planteamiento frecuente es la sustitución de un IFN de baja dosis por otro de mayor dosis y/o frecuencia de administración ante recaídas o actividad lesional en RM. Hay nivel B de evidencia que muestra un beneficio en el aumento de dosis y/o frecuencia, al menos hasta cierto límite. Así, la dosis de 44 μg de IFNβ-1a se mostró ligeramente más eficaz que la dosis de 22 μg del mismo fármaco3; también IFNβ-1b s.c. a días alternos mostró mayor eficacia clínica y en la RM que IFNβ-1a en administración i.m. única semanal38, indicativo de una mejor respuesta al aumentar la dosis y/o la frecuencia; sin embargo, duplicar la dosis de IFNβ-1b a 500 μg no mejoró el beneficio de la dosis estándar de 250 μg39. Así, en sujetos tratados con IFNβ a dosis baja y respuesta insuficiente, la primera opción es aumentar la posología de IFNβ antes de pasar a otro tipo de inmunomodulador, salvo que presenten títulos altos y persistentes de anticuerpos neutralizantes. No deben superarse las dosis máximas autorizadas.
En el segundo grupo, por tratarse de dos fármacos diferentes, hay base teórica para efectuar el cambio en caso de respuesta insuficiente o de intolerancia al tratamiento. La evidencia disponible muestra que el cambio debe hacerse principalmente buscando una mejor tolerabilidad y no una mejor respuesta. Un estudio amplio reciente40 de nivel B de evidencia no mostró diferencia en la respuesta clínica entre IFNβ-1a y AG ni en la tolerabilidad. Sin embargo, hay varios estudios que ofrecen nivel C de evidencia para cambiar de IFNβ por AG en casos de intolerancia o ineficacia, y viceversa. Uno de ellos41 en sujetos con IFNβ-1b que se cambió por AG por toxicidad o ineficacia, con un seguimiento de 3 años, los que cambiaron por ineficacia presentaron una reducción significativa del número de brotes, pero no de la toxicidad. Otro estudio42 comparó los tres cambios posibles en este grupo: de un IFNβ a otro, de AG a un IFNβ y de un IFNβ a AG. Los cambios fueron siempre por control insuficiente de la actividad de la enfermedad, por persistencia de brotes, actividad en la RM o acúmulo de discapacidad; los tres grupos se beneficiaron del cambio de fármaco en cuanto a reducción de brotes, salvo los que cambiaron a AG. Las limitaciones metodológicas lastran el nivel de evidencia, pero parece que en los sujetos con EMRR sin alta actividad o acúmulo de discapacidad asociada a las recaídas, el cambio entre estos inmunomoduladores es una opción segura y razonable.
En el tercer grupo, la experiencia controlada es muy limitada. Dada la pertenencia de la azatioprina a un grupo farmacológico alejado de los inmunomoduladores antes citados, es razonable plantear la sustitución de este fármaco por AG o IFN en caso de intolerancia o ineficacia. Teóricamente, el cambio en el sentido opuesto es también razonable, pero hay que considerar que las evidencias a favor son más tenues, la eficacia esperable puede demorarse meses y que existe cierto riesgo oncogénico.
Escalado terapéuticoEl cambio de un medicamento de primera línea a uno de segunda línea supone entrar en el ámbito del escalado terapéutico. El fundamento del escalado terapéutico es la utilización secuencial de medicamentos con mayor eficacia, aunque también mayor toxicidad, de acuerdo con un esquema preestablecido, en un intento de maximizar el equilibrio riesgo/beneficio, de modo que los medicamentos con mayor toxicidad potencial queden reservados para los pacientes con una enfermedad más agresiva. Los esquemas de escalado terapéutico tienen a su favor la evidencia de clase A para los fármacos aprobados, pero esa evidencia va disminuyendo a medida que se avanza en el escalado. Cuentan con algunos puntos débiles como la definición de respuesta inadecuada, sobre la que no hay un acuerdo definitivo, y con el riesgo de que una aplicación excesivamente esquemática pueda suponer una pérdida de tiempo crítica en algunos casos. Se han publicado varios consensos europeos, de países de lengua alemana y del MS Therapy Consensus Group43-46.
El escalado no se debe hacer buscando una mejor tolerabilidad. Tras agotar las opciones terapéuticas de primera línea, si persiste la falta de respuesta, se debe iniciar la segunda línea de tratamiento, en monoterapia, sin arrastrar fármaco alguno de primera línea. La segunda línea se debe iniciar con natalizumab, dejando mitoxantrona como segunda y última opción, por su toxicidad que limita el periodo de tratamiento. Los criterios de respuesta mencionados antes en la sección correspondiente son de aplicación para decidir pasar a segunda línea o finalizarla y progresar a tratamiento de rescate.
Una alternativa a los esquemas de escalado es la propuesta de terapia de inducción cuyo fundamento teórico sería la consecución rápida de un control de la inflamación para evitar el daño parenquimatoso y la diseminación epitópica. Esto se llevaría a cabo mediante el empleo inicial de un medicamento inmunosupresor potente durante un tiempo limitado, seguido del uso de un inmunomodulador como mantenimiento. Hay experiencia limitada con mitoxantrona seguida de interferón o acetato de glatirámero47,48, pero en la actualidad no hay base sólida como para recomendar esta opción.
Finalmente, la falta de información suficiente sobre la eficacia de la terapia combinada no hace posible asignarle un papel concreto dentro del esquema de escalado terapéutico.
Terapias de rescateTratamiento de rescate es el empleo de fármacos no autorizados en EM, pero sí en otras enfermedades autoinmunitarias, o bien la instauración de combinaciones terapéuticas de fármacos autorizados como monoterapia en la EM. La elección del tratamiento de rescate depende de la experiencia y el criterio del neurólogo. Por razones de seguridad se preferirán los fármacos más probados en otras enfermedades. La monoterapia con un fármaco no autorizado para la EM es preferible a la combinación de dos fármacos autorizados, porque la probabilidad de sinergia es remota y es probable un incremento de la toxicidad.
Propuesta de escalado terapéutico en la EM
- 0.
Tratamiento de primera línea:
- •
IFNβ-1b s.c., IFNβ-1a s.c. o i.m.
- •
Acetato de glatirámero.
- •
Azatioprina.
- •
- 1.
Persistencia de brotes con escasa o nula repercusión sobre el estado funcional:
- •
Si IFN: aumentar la frecuencia o la dosis.
- •
Si IFN de alta dosis y frecuencia: valorar cambio a AG.
- •
Si AG: valorar cambio a IFN.
*En caso de persistir los brotes, tras haberse realizado las anteriores opciones, se puede valorar la asociación de azatioprina (ésta no es una opción refrendada por estudios controlados, aunque practicada por algunos neurólogos; véase apartado de azatioprina).
- •
- 2.
Persistencia de brotes con incremento significativo de la discapacidad, opciones terapéuticas de los puntos anteriores llevadas a cabo. Pacientes con comienzo agresivo: recaídas muy seguidas con incremento importante de discapacidad:
- •
Natalizumab.
- •
- 3.
Pacientes con intolerancia a natalizumab o con baja respuesta terapéutica a este fármaco:
- •
Mitoxantrona.
- •
- 4.
Pacientes con tratamientos inmunosupresores previos sin control de la actividad:
- •
Natalizumab: esperar tiempo suficiente para tener evidencias de inmunocompetencia antes de iniciar natalizumab.
- •
En caso de falta de respuesta a natalizumab, considerar el uso compasivo de: rituximab, alemtuzumab o ciclofosfamida.
- •
- 5.
En los dos puntos anteriores, una vez lograda la remisión de la enfermedad o el agotamiento de la dosis de inmunosupresor, se puede valorar la vuelta a la terapia inmunomoduladora con un agente diferente del empleado previamente.
- 6.
Ninguna respuesta a tratamientos previos: valorar trasplante de médula ósea (fig. 1).
.")
El tratamiento con anticuerpos monoclonales e inmunosupresores está contraindicado durante el embarazo y la lactancia. En general, el tratamiento con inmunomoduladores se desaconseja durante el embarazo y la lactancia.
En pacientes embarazadas que presentan una alta tasa de brotes antes de iniciar el tratamiento, el riesgo de aparición de un brote grave tras la interrupción del tratamiento con interferón beta debe tenerse en cuenta frente al posible riesgo de un aborto espontáneo. Se debe informar a la paciente que se quede embarazada o que esté planificando un embarazo mientras está en tratamiento de los riesgos potenciales y debe considerarse la posibilidad de interrumpir el tratamiento.
En principio, la opción de administrar IVIG debe considerarse restringida. Las mujeres en edad fértil deben utilizar medidas anticonceptivas apropiadas. Se permite el empleo de IVIG para el tratamiento de los brotes durante el embarazo, y de los corticoides a partir del segundo trimestre. Hay autores que recomiendan iniciar el tratamiento inmunomodulador inmediatamente después del parto para evitar la aparición de brotes durante el puerperio, mientras otros permiten la lactancia materna iniciando IVIG tras el parto.
Durante la lactancia se desaconseja el uso de inmunosupresores, anticuerpos monoclonales e inmunomoduladores para evitar posibles efectos adversos en el lactante.
Uso pediátricoLos IFNβ y AG pueden administrarse a niños con EM, a partir de los 12 años de edad. Cuando se disponga de más de una dosis, se administrará la más baja. Se dispone de una información limitada sobre el uso de inmunomoduladores en niños menores de 12 años, restringiéndose su uso a casos particulares. Mayores precauciones deberían tomarse en el caso de uso compasivo de inmunosupresores. La experiencia con natalizumab en niños que no responden a inmunomoduladores es muy limitada49.
EpílogoDebido a los avances en las técnicas diagnósticas y la inminente llegada de nuevos fármacos, es previsible que las recomendaciones de este consenso deban ser revisadas periódicamente.

