



Primary immunodeficiency diseases (PIDs) are disorders associated mainly with recurrent and severe infection and an increase in susceptibility to autoimmune conditions and cancer. In Venezuela, PIDs are underdiagnosed and there is usually a delay in their diagnosis. Hence there are no data concerning the frequency and type of PIDs that occur. The aim of this study was to identify and quantify the types of PIDs that occur in Merida, a population within Venezuela.
Patients or materials and methodsFollowing an informative program designed to alert local health professionals to the warning signs for PIDs, patients with a history of recurrent infections were referred to the Instituto de Inmunologia Clinica, Universidad de Los Andes.
Results and conclusionsDuring the three-year period January 2014 to January 2017, thirty-two cases of PIDs were identified in pediatric patients, and 17 different types of PIDs, were identified. Predominantly antibody deficiencies were most frequent (40.6%), followed by immunodeficiencies affecting cellular and humoral immunity (21.8%), congenital defects of phagocyte (18.7%), CID with associated or syndromic features (9.3%), defects in intrinsic and innate immunity (6.4%) and diseases of immune dysregulation (3.2%). These results have important implications not only to the future approach for management of patients in our regions, but add important knowledge concerning PIDs in Latin America and worldwide.
Primary immunodeficiency diseases (PIDs) are disorders associated mainly with recurrent and severe infections, and an increase in susceptibility to autoimmune conditions and cancer.1 PIDs are still frequently undiagnosed or diagnosed late, since in many cases mild-to-moderate symptoms might not arouse physicians’ suspicion, or patients with severe forms may die at an early age due to systemic infections and subsequent complications without ever being properly diagnosed.2 Hence, it is now accepted that the prevalence of PIDs is underestimated,3 and more common than generally thought.4
Early diagnosis of PIDs is crucial for improving the quality of life in patients. A delay in diagnosis, or inadequate treatment, results in increased mortality and morbidity in affected individuals, and is associated with complications, including resistance to treatment and the association of unusual causative microorganisms.5 Depending on the affected component of the immune system, infections may vary from mild, to severe life-threatening systemic conditions.6,7 To date, mutations in about 350 genes have been identified in PIDs, the criteria have been updated and have been classified into nine different categories: immunodeficiencies affecting cellular and humoral immunity, predominantly antibody deficiencies, diseases of immune dysregulation, congenital defects of phagocyte number, function, or both, defects in Intrinsic and Innate immunity, auto-inflammatory disorders, complement deficiencies, and phenocopies of PID.8 There are correlations between the nature of the infectious agents (such as viral, extracellular and intracellular bacterial or fungal), and the type of deficiencies, which are also related to the patient's genetics. In general, the more severe deficiencies result in infections that present early in infancy.9
The frequency in different countries is variable, affecting one in 1200 live births, worldwide.10 Diagnosis of PIDs is still a challenge in many countries, especially in Latin America (LA), which face social and economic problems. Registry reports of several countries show wide variations in geographical prevalence. Argentina has the highest number of registered cases (2323 patients), with a frequency of approximately one patient per 20,000 citizens. Brazil has the second largest number of recorded cases and has a frequency of one patient per 146,000 inhabitants. Other countries such as: Costa Rica (1/48,000), Colombia (1/51,000), Mexico (1/98,000), Uruguay (1/121,000), Honduras (1/145,000), Chile (1/165,000), Paraguay (1/350,000), and Peru (1/516,000) also displayed variable frequency.11 In Venezuela, PIDs are undiagnosed or diagnosed late, and there are no data about the frequency and type of PIDs. Only a few studies have been published and reported up to 2007,12–14 showing the lowest incidence in Latin American countries between 0.02 and 0.05 cases per 100,000 births.14 The perspective for PIDs patients in Latin America, is limited by a low diagnosis rate, scarce specialized clinics, poor knowledge among clinicians and inaccessibility for diagnostic tests, and treatments.15 PIDs represent a public health problem, not only because they are not as rare as commonly believed, since cumulatively they have a global incidence of 10.3 per 100,000 person-years,4 but also because of their high cost to the health system. Timely diagnosis is crucial for improving the quality of life in patients with PIDs, and the outcome of the disease can be favorably affected by increasing physician awareness and the establishment of specific laboratory facilities.16
Thus, this work was aimed to evaluate the immune response in pediatric patients with recurrent and severe infection to detect the presence of PIDs in Merida, a city in Venezuela, in order to determine the prevalence and the kind of PIDs in our region. To achieve this goal, patients who were referred because of recurrent infections and who fulfilled specific criteria were evaluated according to a predefined examination, including a set of laboratory analyses. In total, 32 children with PIDs, were identified during a three-year period in pediatric patients, and we were able to identify 17 different types of PIDs, in the study patients. These results have an important implication not only for the future approach to management of patients in our local region, but also add important knowledge concerning PIDs in Latin America and worldwide.
Material and methodsPatients’ enrollmentThis study was conducted in accordance with the Helsinki declaration, with informed consent obtained from each patient's family. The local ethics committee of the Universidad de Los Andes approved the study. Initially, an informative program was implemented, designed for health professionals (primary care physicians, pediatricians, internists, students, and residents). Every four months, printed warning signs flyers were distributed among physicians and students. The study was performed in Merida state, which has an estimated population of 1,025,445 with a population density of 90.74hab/km and an average annual birth rate for 2017 of 18.8 births per 1000 people in the population. The children in the study were referred to the Instituto de Inmunología Clínica, Facultad de Medicina, Universidad de Los Andes, by either general physicians or pediatricians, when a history of recurrent infections was documented. During a period of three years, from January 2014 to January 2017, the children were evaluated according to a predefined examination schedule. The inclusion criteria in the study, were based on the Warning Signs.2 The initial studies including immunoglobulin serum levels, leucocytes count, platelets count, subpopulation characterization and complement component, were carried out.17 Additional functional and molecular studies were performed according to initial finding.
ReagentsComplete media consisted of RPMI 1640 supplemented with 2mM L-glutamine, 10% FCS, 100U/ml penicillin, and 100μg/ml streptomycin (all reagents purchased from GIBCO, Life Technologies, Inc.) anti-human CD40 mAb-FITC, anti-human CD40 ligand-PE, anti-human CD4-FITC/CD8-PE/CD3-PerCp, anti-human CD3-FITC/CD19-PE/CD45-PerCp, CD56/CD16-PE/CD3-FITC, anti-CD4-FITC, anti-HLA-DR-FITC, anti-Ki-67-PerCp, anti-IL-12-PE, anti-INF-g.PE, anti-Foxp3, anti-CD11a-FITC, anti-CD11b-PE, anti-CD11c-PE and anti-CD18-PE were purchased from PharMingen (San Diego, CA, USA). Phorbol 12-myristate-13acetate (PMA) was purchased from Calbiochem (San Diego, CA, USA). Dihydrorhoda- mine-123 (DHR-123) was purchased from Molecular Probes (Eugene, OR, USA). Mouse IgG1 FITC-conjugated and PE were purchased from Becton Dickinson & Co. (San José, CA, USA)
Peripheral blood mononuclear cells (PBMCs) and polymorphonuclear cells (PMNs) purificationPBMCs were isolated from heparinized blood by density gradient sedimentation over Lymphoprep. Freshly isolated PBMCs (98% viable evaluated by trypan blue exclusion) were resuspended in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 100units/ml of penicillin and 100μg/ml of streptomycin. For PMNs, heparinized blood was mixed with 6% dextran solution (mol wt 500,000) and incubated at room temperature for 20min. Leukocyte-enriched supernatant was collected, diluted with RPMI 1640, and layered on Ficoll-Hypaque (1077, Sigma, St. Louis, MO, USA). After density gradient centrifugation at 400×g for 30min, PMNs were obtained from the bottom. Red blood cells contained in the PMNs pellet were hypotonically lysed for 30s using cold distilled water. Purified PBMC and polymorphonuclear cells (98% viable) were resuspended at 2×106cells/ml in RPMI supplemented with 10% FBS.
Subpopulation and cell adhesion molecules (CAMs) identificationComplete blood counts were measured using standard laboratory methods. Analysis of lymphocyte subpopulations defined by expression of CD3 (T cells), CD4 (T helper cells), CD8 (T cytotoxic cells), CD19 (B cells) and CD16/56 (NK cells). PMNs populations were also labeled by using CD11a, CD11b, CD11c and CD18, and analyzed by flow cytometry. Three-color flow cytometry was performed by FACSort. Cells were incubated with antibody mixture during 30min, and after three washes were analyzed by flow cytometry (Becton Dickinson, San Jose).
PBMCs activation and proliferationPBMCs were plated in tissue culture plates with complete media with or without (untreated control) Phytohaemagglutinin (PHA) (20μg/ml) or Phorbol 12-myristate 13-acetate (PMA) (100ng/ml). Cells were incubated in a humidified atmosphere with 5% CO2, for 48h for proliferation assays or six hours for activation or cytokines induction.
Superoxide productionFlow cytometric analysis of neutrophil respiratory burst activity was performed. Briefly, freshly isolated neutrophils (1×106cells/ml) were preloaded with DHR-123 (1mmol/L) at 37°C for 15min. Afterwards, 100ng/ml PMA was added and incubated for 20min at 37°C. Cells were analyzed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). A total of 10,000 events were collected from each sample.
Western blotCells at a concentration of 2×106/100μl, were lysed on ice for 15min in 1ml of a buffer containing: 50mM Tris–HCl pH 7.4; 150mM NaCl, 1mM ethylenediaminetetraacetic acid (EDTA), 1mM phenylmethylsulfonyl fluoride (PMSF), 1μg/ml leupeptin/aprotinin, 1mM sodium orthovanadate and centrifuged at 14,000×g for 10min to yield post-nuclear cell lysates. Proteins were solubilized in SDS sample buffer separated by SDS–PAGE, transferred to Polyvinylidene difluoride (PVDF) membrane and probed with corresponding primary antibodies (anti-ZAP70, anti-p56lck, anti-ICOS, anti-foxp3, anti-p22-phox, anti-CD3ζ, CD3ɛ, LAT anti-p47-phox, anti-p67-phox, anti-gp91-phox, anti-ERK) and secondary antibody conjugated to HRP. Blots were visualized by chemiluminescence, by using the enhanced chemiluminescence (ECL) system from Pierce, Rockford, IL, USA.
RNA isolation RT-PCRRNA was extracted and stored at −80°C for additional genes analysis. RT-PCR amplification was carried out in a conventional thermocycler or Chromo4™ (BioRad), using SYBR Green master mix (primers used to molecular for assays are listed in Table 1). After a preincubation step at 95°C for 15min, the amplification was performed for 40 cycles including denaturation (94°C/15s), annealing (60°C/20s) and extension (72°C/25s). For Real time PCR a melting curve analysis was performed on the products by heating from 72°C to 95°C, a single fluorescent signal was obtained each 0.2°C.
RT-PCR primers.
| Molecule | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| APRIL | TCTCCTTTTCCGGGATCTCT | CCAGAATGGGGAAGGGTATC |
| BAFF-R | AGGACGCCCCAGAGC C | AGTGTCTGTGCTTCTGCAGG |
| BLNK | GCAAGACACTTCCCAGTAA | CTTGTCTGTGACTTGACCC |
| BTK | CAGGCTGAGCAACTGCTAA | GGAGCCTTCTTTGATCATC |
| CD19 | TCGCTTTCTTTTCCTCCTCA | TACTATGGCACTGGCTGCTG |
| CD40 | TGCCAGCCAGGACAGAAACT | GGGACCACAGACAACATCAG |
| CD40L | ACTTTTTGCTGTGTATC | CGGAACTGTGGGTATTT |
| DOCk8 | GAACAGCCTGGATGTGCAGCTTGCCC | TTAGCTGCCCTGTGACAACTGGGTTTC |
| Common gamma (γc) | ACGGGAACCCAGGAGACAGG | AGCGGCTCCGAACACGAAAC |
| GAPDH | CTCTGCTCCTCCTGTTCGAC | ACGACCAAATCCGTTGACTC |
| IL-7Rα | CCATGTTTCTTTTAGGTATAT | GTGCCCATCAAAATTTTATTC |
| SAP | GCCTGGCTGGCGTAGCAGGGGCATCTCC | ATGTACAAAAGTCCATTTCAGCTTTGAC |
| STAT3 | TGCGGCAGTTTCTGGCCCCTTGGATTG | TCCTCTGAGAGCTGCAACGTCCCCAG |
| Tap1 | GGTGTCCCGCTCCCCGCGGGTGCCG | CCCTCCCGGATAGCGCCTCCTTCCA |
| Tap2 | CCTGGACCTCCCTGCTGCTGGTGGA | CAGGGCCTGCTCGCACTGCACATCTAG |
| TLR3 | TTAAAGAGTTTTCTCCAGGGTGTTT | AATGCTTGTGTTTGCTAATTCCA |
| TYk2 | GCCTGAAGGAGTATAAGTTC | TGAGATGATAGACCTCACAG |
| XIAP | AAGAGAAGATGACTTTTAA | TGCTGAGTCTCCATATTGC |
Data were collected in Excel database and were converted for analysis using the Prism software Version 5.03.
ResultsA total of 60 patients were referred to Instituto de Inmunología Clínica Facultad de Medicina, Universidad de Los Andes (Idic-ULA), during January 2014 to January 2017. The examination schedule consisted of medical history, physical examination and a set of laboratory analyses. 32 patients met the inclusion criteria. The remaining patients were excluded from the study because they did not meet the PIDs criteria. Patients were admitted for recurrent infections, and the main presenting symptoms in 13 cases (40.6%) were upper and lower respiratory infection, followed by seven cases (21.9%) of skin abscesses, six cases (18.8%) of chronic diarrhea and two cases (6.3%) of encephalitis. The most common first presentation of PIDs patients was pneumonia, observed in 16 cases (50%). It was followed by diarrhea in six cases (18.8%), omphalitis and/or delayed umbilical cord detachment in two cases (6.3%), otitis media in six cases (18.8%), recurrent sinusitis in nine cases (28.1%), abscesses in seven cases (21.9%), lymphadenopathy in three cases (9.4%), mucocutaneous candidiasis in one case (3.1%), BCGosis in one case (3.1%), encephalitis in two cases (6.2%), and hepatosplenomegaly in one case (3.1%). There was a single observation of a rare PID complication in a patient affected by SCID, which was diagnosed as Kawasaki disease.
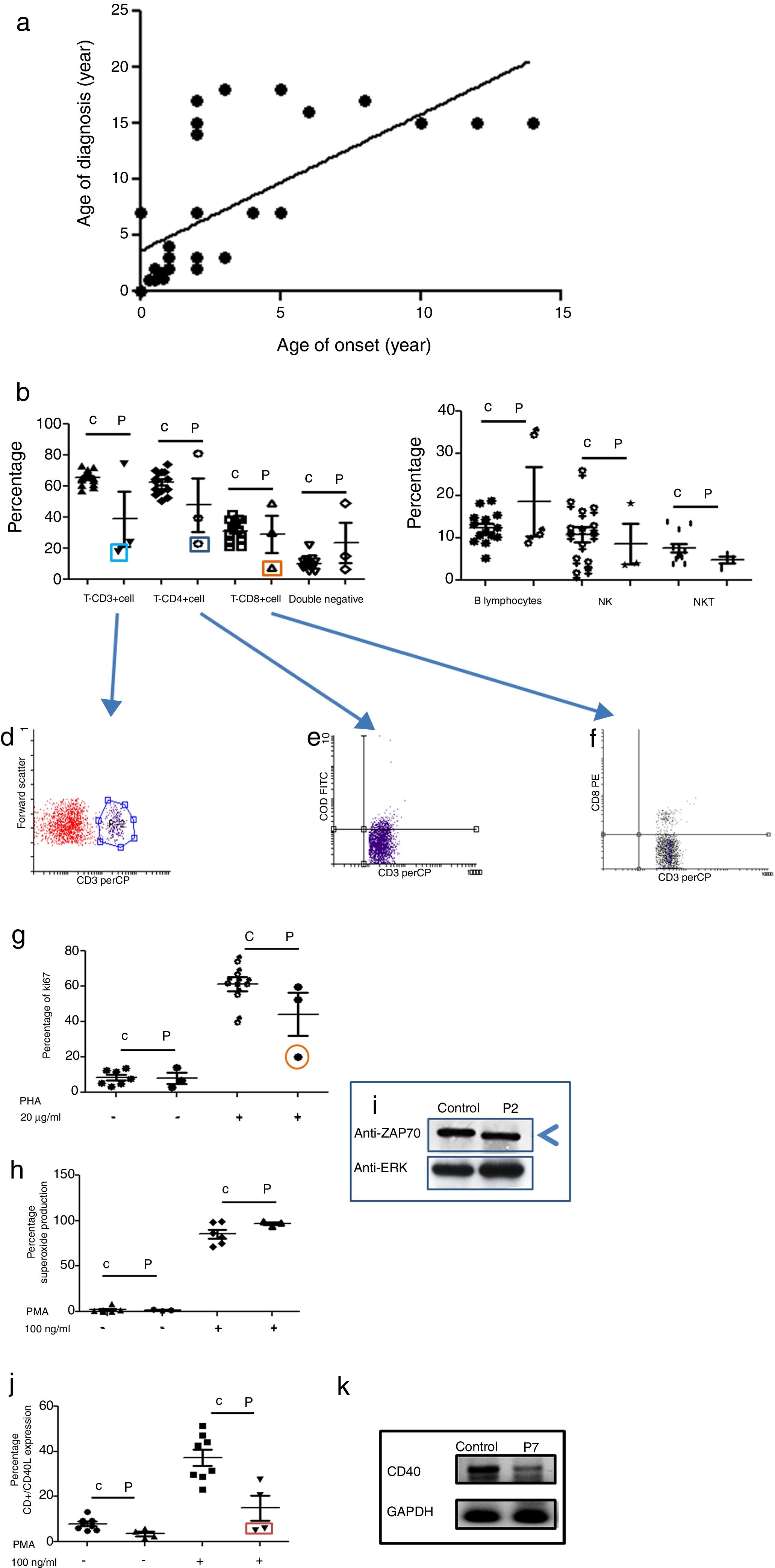
Age of onset, age of diagnosis, and diagnosis delayThe age of onset of symptoms ranged between 0 and 16 years with a mean of 4.7 years for all PID categories. The age of the patients at the time of diagnosis ranged between 0.01 and 16 years with a mean of 7.3 years. The majority of the patients (72%) were diagnosed during childhood (<14 years). The overall mean delay in diagnosis was 4.32 years, in which patients suffered from one to 20 infections (see Fig. 1a); this is a longer diagnosis delay in comparison to other reports.18,19
. 1h. Represents superoxide production in PMNs, before and after stimulation (PMA 100ng/ml). 1i. Shows a ZAP70 western blot in control and patient with CD8 deficiency. 1j. Shows CD4+CD40L expression in controls and Hyper-IgM patients. 1k. Shows RT-PCR of CD40 in control and Hyper-IgM patient.")
Phenotypic and functional analysis of leukocytes in patients with immunodeficiencies affecting cellular and humoral immunity. 1a. Shows the relationship between age onset and age of diagnosis. 1b. Shows T cell subpopulation in controls and combined-T cell deficiency patients. 1c. Shows B cell and NK-NKT subpopulation in controls and combined-T cell deficiency patients. Square light blue and 1d. Graphic represents a patient with CD3 deficiency. Square dark blue and 1e. Graphic represents a patient with CD4 deficiency and Square orange and 1f. Graphic represents a patient with CD8 deficiency. 1g. Represents proliferation assays measured by ki67 expression in T lymphocytes, before and after stimulation (PHA 20μg/ml). 1h. Represents superoxide production in PMNs, before and after stimulation (PMA 100ng/ml). 1i. Shows a ZAP70 western blot in control and patient with CD8 deficiency. 1j. Shows CD4+CD40L expression in controls and Hyper-IgM patients. 1k. Shows RT-PCR of CD40 in control and Hyper-IgM patient.
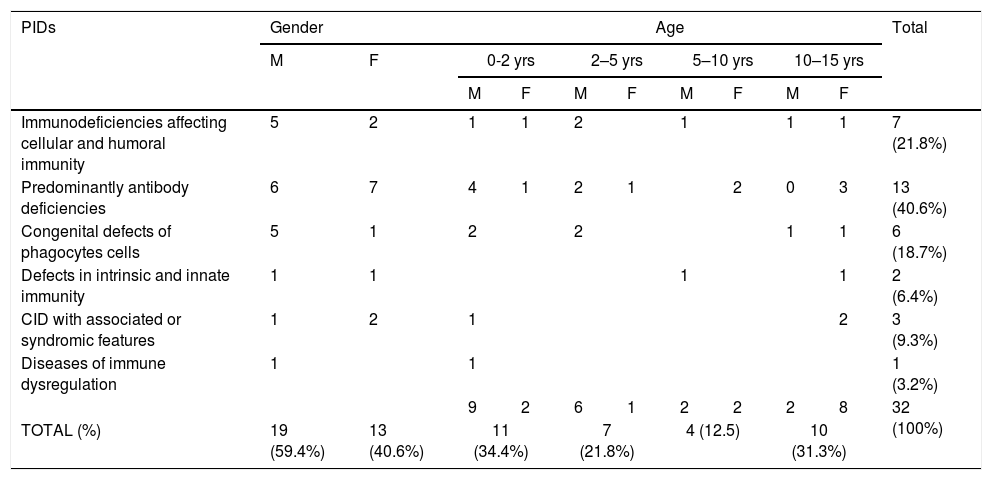
A total number of 32 PIDs patients: 19 male (59.4%) and 13 female (40.6%), with a median age of seven years (Range 1m–17 years), were enrolled and 17 types of PIDs were identified in the study population. Predominantly antibody deficiencies PIDs were the most common group of PIDs affecting 13 patients (40.6%), followed by immunodeficiencies affecting cellular and humoral immunity affecting seven patients (21.8%), congenital defects of phagocytes cells affecting six patients (18.7%), CID with associated or syndromic features affecting three patients (9.3%), defects in intrinsic and innate immunity affecting two patients (6.4%), and PIDs associated with diseases of immune dysregulation affecting one patient (3.2%) (see Table 2).
PIDs diagnosis and frequency.
| PIDs | Gender | Age | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M | F | 0-2 yrs | 2–5 yrs | 5–10 yrs | 10–15 yrs | ||||||
| M | F | M | F | M | F | M | F | ||||
| Immunodeficiencies affecting cellular and humoral immunity | 5 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 7 (21.8%) | ||
| Predominantly antibody deficiencies | 6 | 7 | 4 | 1 | 2 | 1 | 2 | 0 | 3 | 13 (40.6%) | |
| Congenital defects of phagocytes cells | 5 | 1 | 2 | 2 | 1 | 1 | 6 (18.7%) | ||||
| Defects in intrinsic and innate immunity | 1 | 1 | 1 | 1 | 2 (6.4%) | ||||||
| CID with associated or syndromic features | 1 | 2 | 1 | 2 | 3 (9.3%) | ||||||
| Diseases of immune dysregulation | 1 | 1 | 1 (3.2%) | ||||||||
| 9 | 2 | 6 | 1 | 2 | 2 | 2 | 8 | 32 (100%) | |||
| TOTAL (%) | 19 (59.4%) | 13 (40.6%) | 11 (34.4%) | 7 (21.8%) | 4 (12.5) | 10 (31.3%) | |||||
M: male, F: female.
Immunodeficiencies affecting cellular and humoral immunity, comprised 21.8% of the patients, being evident in seven patients: five male, and two female. The average age of disease onset was 30.5 months. These patients experienced severe infections in different organs and systems, requiring multiple hospitalizations, treatment with broad-spectrum antimicrobial therapy, and were demonstrated to be infected by multiple infectious agents including viruses, extracellular and intracellular bacteria. Phenotypic and functional characterization demonstrated a deficiency of T-CD3 lymphocytes (T-CD3+), associated to low serum immunoglobulins in combined immunodeficiency, T-CD4 cell deficiency, and T-CD8 lymphocyte deficiency (T-CD8) (see Table 3 and Fig. 1b–f). In all three cases, B and NK lymphocyte populations were conserved (see Fig. 1c). Functional tests showed low proliferative response only in the combined immunodeficiency patient, with a CD3+T lymphocyte defect (see Fig. 1g); while superoxide production was preserved in all of three cases (see Fig. 1h). CD3+T lymphocytes deficiency is associated with IL-7Rα and ζ chain genes mutations, among others.20 We performed RT-PCR assays to detect IL-7Rα and ζ chain genes expression, but none of them were affected in our patient (data not shown). The CD4 deficient patient died during evaluation, and no samples for molecular assays could be obtained. CD8+T lymphocytes deficiency is associated with TAP1/2, lck and Zap70 genes mutations.21 In our case, a low molecular weight of Zap70 protein was detected (see Fig. 1i). In the patients identified with Hyper IgM syndrome, two male patients were shown to not express CD40L (Fig. 1j), one female patient was associated with reduced expression of CD40 (Fig. 1k), and the other male patient died during the study.
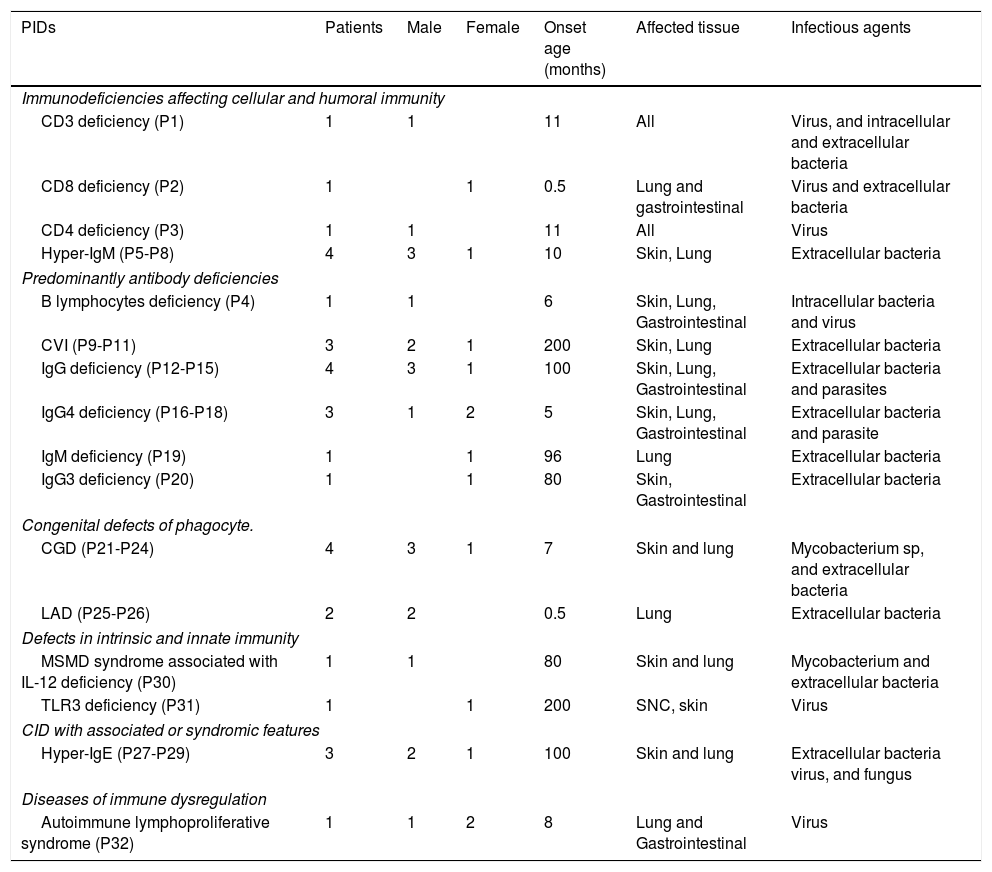
PIDs characteristic.
| PIDs | Patients | Male | Female | Onset age (months) | Affected tissue | Infectious agents |
|---|---|---|---|---|---|---|
| Immunodeficiencies affecting cellular and humoral immunity | ||||||
| CD3 deficiency (P1) | 1 | 1 | 11 | All | Virus, and intracellular and extracellular bacteria | |
| CD8 deficiency (P2) | 1 | 1 | 0.5 | Lung and gastrointestinal | Virus and extracellular bacteria | |
| CD4 deficiency (P3) | 1 | 1 | 11 | All | Virus | |
| Hyper-IgM (P5-P8) | 4 | 3 | 1 | 10 | Skin, Lung | Extracellular bacteria |
| Predominantly antibody deficiencies | ||||||
| B lymphocytes deficiency (P4) | 1 | 1 | 6 | Skin, Lung, Gastrointestinal | Intracellular bacteria and virus | |
| CVI (P9-P11) | 3 | 2 | 1 | 200 | Skin, Lung | Extracellular bacteria |
| IgG deficiency (P12-P15) | 4 | 3 | 1 | 100 | Skin, Lung, Gastrointestinal | Extracellular bacteria and parasites |
| IgG4 deficiency (P16-P18) | 3 | 1 | 2 | 5 | Skin, Lung, Gastrointestinal | Extracellular bacteria and parasite |
| IgM deficiency (P19) | 1 | 1 | 96 | Lung | Extracellular bacteria | |
| IgG3 deficiency (P20) | 1 | 1 | 80 | Skin, Gastrointestinal | Extracellular bacteria | |
| Congenital defects of phagocyte. | ||||||
| CGD (P21-P24) | 4 | 3 | 1 | 7 | Skin and lung | Mycobacterium sp, and extracellular bacteria |
| LAD (P25-P26) | 2 | 2 | 0.5 | Lung | Extracellular bacteria | |
| Defects in intrinsic and innate immunity | ||||||
| MSMD syndrome associated with IL-12 deficiency (P30) | 1 | 1 | 80 | Skin and lung | Mycobacterium and extracellular bacteria | |
| TLR3 deficiency (P31) | 1 | 1 | 200 | SNC, skin | Virus | |
| CID with associated or syndromic features | ||||||
| Hyper-IgE (P27-P29) | 3 | 2 | 1 | 100 | Skin and lung | Extracellular bacteria virus, and fungus |
| Diseases of immune dysregulation | ||||||
| Autoimmune lymphoproliferative syndrome (P32) | 1 | 1 | 2 | 8 | Lung and Gastrointestinal | Virus |
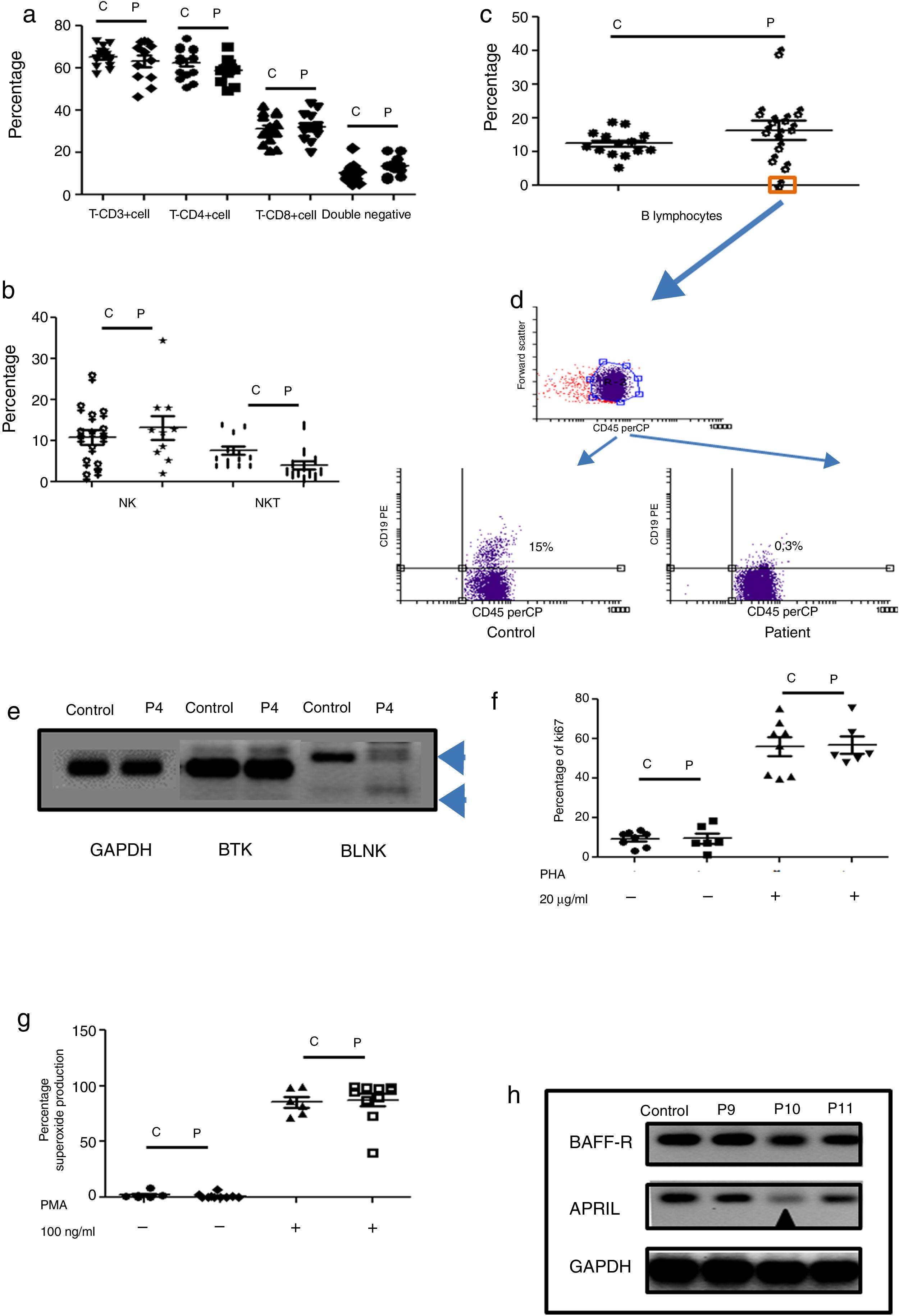
Humoral immunodeficiencies were the most frequent in the studied group (see Table 2). These immunodeficiencies may result from the absence or decrease of B lymphocytes in peripheral blood or from defects in the activation of their effector functions associated with immunoglobulin isotype change, maturation of antibody affinity or defects in population development memory effectors.22 There were a total of 13 such patients, six male, and seven female, with an average age of onset of the disease of 66 months in the study group. Respiratory, skin, and soft tissue infection were present, and extracellular bacteria were predominant (see Table 3). Only one male patient had defects in B lymphocytes development (see Table 3 and Fig. 2c (orange box), and 2d), with the absence of circulating B cells. T cell and NK/NKT were not affected (see Fig. 2a and b). Both proliferative response and superoxide production, were conserved (Fig. 2f–g). The absence of B lymphocytes is associated with mutations in genes involved in development, within which BTK, BLK, μ chain are prominently involved. In our case, the patient showed an absence in the BLNK, which explains the ontogeny of this defect (Fig. 2e). The other patients showed functional defects in the activation and differentiation of B lymphocytes, for different causes: common variable immunodeficiency (CVID) in three cases, total IgG defect in four cases, IgG4 subclass defect in three cases, IgG3 subclass defect in one case, IgM defect in one case (Table 3). Additionally, BAFF-R and APRIL expression was evaluated in patients with CVID and a marked reduction of APRIL was observed in one patient (see Fig. 2h).
. 2g. Represents superoxide production in PMNs, before and after stimulation (PMA 100ng/ml). 2h. Shows RT-PCR of BAFF-R and APRIL in control and CVI patients.")
Phenotypic and functional analysis of PBMCs in patients with defects associated predominantly antibody deficiencies. 2a. Shows T cell subpopulation in controls and patients with humoral deficiency. 2b. Shows NK/NKT subpopulation in controls and patients with humoral deficiency. 2c. Shows B cell subpopulation in controls and patients with humoral deficiency. Square orange and 2d. the graphic represents a patient with B cell deficiency. 2e. Shows RT-PCR of BTK and BLNK in control and patient with B cell deficiency. 2f. Represents proliferation assays measured by ki67 expression in T lymphocytes, before and after stimulation (PHA 20μg/ml). 2g. Represents superoxide production in PMNs, before and after stimulation (PMA 100ng/ml). 2h. Shows RT-PCR of BAFF-R and APRIL in control and CVI patients.
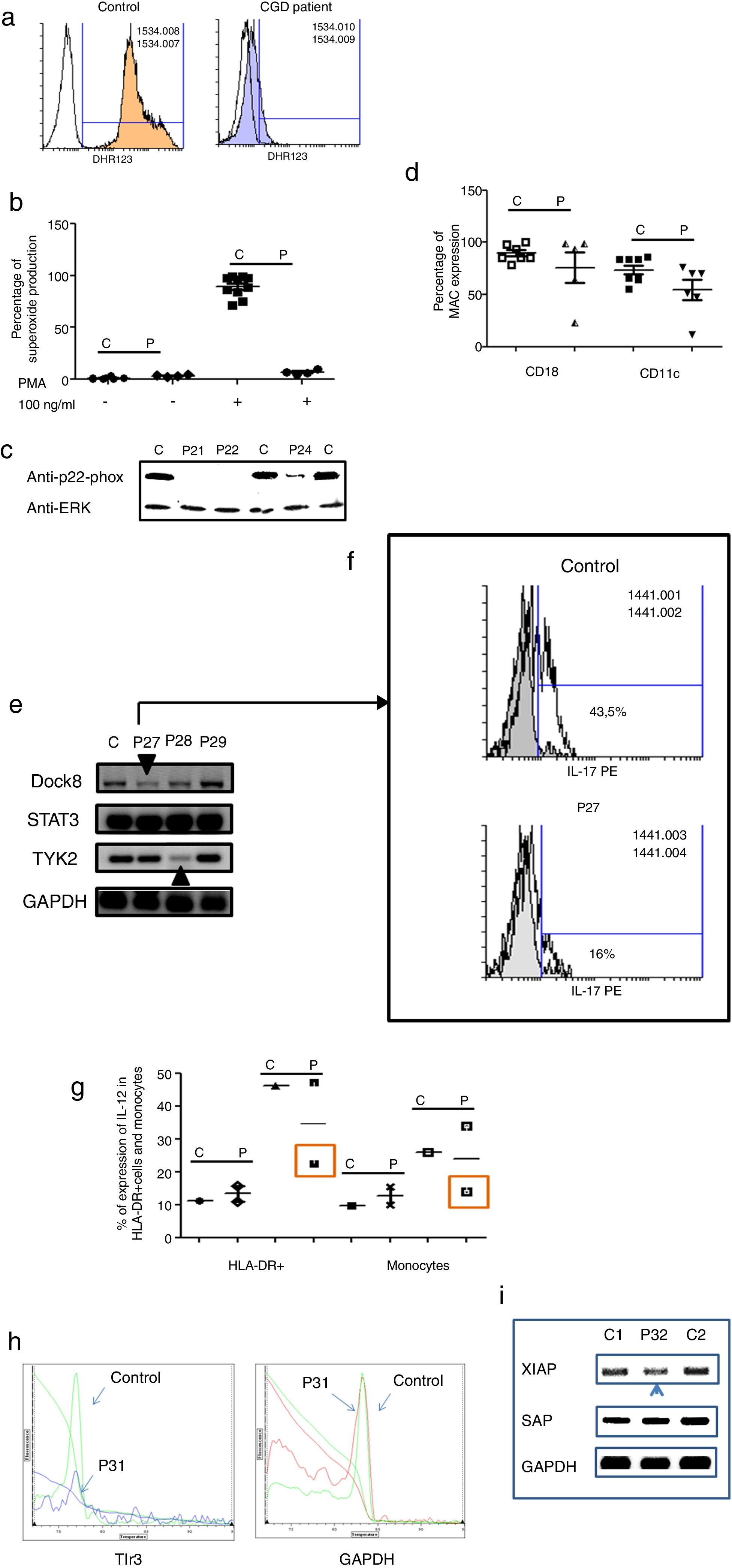
Three male and one female patients presented with Chronic Granulomatous Disease, as evidenced by their failure to produce superoxide (see Fig. 3a–c), and of these, three also exhibited p22-phox deficiency. The fourth patient died before molecular studies could be performed. Two patients presented with delayed umbilical cord detachment of, on average, 47.5 days. In addition, they experienced complicating respiratory infections by extracellular bacteria, omphalitis, and required hospitalization (see Table 3). These patients showed CD18 and CD11c deficiency associated with leukocyte adhesion deficiency (LAD) (See Fig. 3d).
. 3c. Shows a p22-phox western blot in controls and CGD patients. 3d. Shows CD18 (left panel) and CD11c (right panel) in LAD patients. 3e. Shows RT-PCR of DOCK8, STAT3 and TYK2 in control and Hyper-IgE patients. 3f. Shows IL-17 production in control and P27. 3g. Shows IL-12 production by HLA-DR+ cells (left panel) and monocytes (right panel) in controls and MSMD syndrome patients. 3h. Shows TLR3 expression in controls and patient, measured by real-time PCR. 3i. Shows RT-PCR of XIAP and SAP in controls and patient with autoimmune lymphoproliferative syndrome.")
Analysis of leukocytes in patients with defects associated with other PIDs: Defects in intrinsic and innate immunity, CID with associated or syndromic features and Diseases of immune dysregulation. 3a. Graphic represents superoxide production in a control and CGD patient. 3b. Represents superoxide production in PMNs from controls and CGD patients, before and after stimulation (PMA 100ng/ml). 3c. Shows a p22-phox western blot in controls and CGD patients. 3d. Shows CD18 (left panel) and CD11c (right panel) in LAD patients. 3e. Shows RT-PCR of DOCK8, STAT3 and TYK2 in control and Hyper-IgE patients. 3f. Shows IL-17 production in control and P27. 3g. Shows IL-12 production by HLA-DR+ cells (left panel) and monocytes (right panel) in controls and MSMD syndrome patients. 3h. Shows TLR3 expression in controls and patient, measured by real-time PCR. 3i. Shows RT-PCR of XIAP and SAP in controls and patient with autoimmune lymphoproliferative syndrome.
The Hyper IgE syndrome (HIgE) were detected in three cases, Mendelian susceptibility to mycobacterial disease (MSMD) in one case and susceptibility to Herpes virus associated to low expression of TLR3 in one case. Hyper-IgE syndrome has been described as associated with mutations in several genes, such as STAT3, TYk2, DOCk8, among others.23 In this work, STAT3, TYk2, DOCk8 were analyzed (see Fig. 3e), and two defects of Dock8 (P27) and TYK2 (P28) were identified. In addition, low IL-17 production was observed in P27, a finding associated with candidiasis mucocutaneous, presented by this patient (Fig. 3f).
Additionally, one patient presented BCGosis, and extrapulmonary infection by Mycobacteria tuberculous at one month of age. This was followed by infections in the skin and soft tissues by extracellular bacteria and in the lower respiratory tree (see Table 3). There was a deficiency in IL-12 production in both monocytes and HLA-DR+ cells. This corresponds to Mendelian Susceptibility to Mycobacterial Disease, (MSDD) and is still under study in order to determine the specific mutation involved. (see Fig. 3g). Moreover, a low expression of TLR3 was identified in one female patient, with encephalitis associated with herpes simplex virus type 1 (HSV-1) (Fig. 3h).
Finally, one patient presented severe EBV and CMV infection, associated with hepatosplenomegaly, lymphadenopathies and lymphocytosis. This type of PIDs has been described as related to mutations in several genes, such as XIAP and SAP. The initial characterization indicates the presence of autoimmune lymphoproliferative syndrome, associated with low expression of XIAP (P32) (see Fig. 3i).
DiscussionThe foremost challenges concerning PIDs diagnosis in our country are the lack of knowledge of PIDs among the health professionals, the few available specialized centers, and the lack of laboratories that are able to carry out the specialized lab tests needed to diagnose these conditions. Because of the importance of early diagnosis and proper treatment in order to prevent complications and premature deaths, we started an educational program directed toward health professionals which would lead them to refer possible PID patients for better evaluation of their disease. This effort resulted in the referral of 60 patients who had experienced recurrent and severe infections, during a three-year period. Of these, a total number of 32 PIDs patients, were enrolled and 17 types of PIDs were identified in the study population. Predominantly antibody deficiencies PIDs were the most common group of PIDs (40.6%), followed by immunodeficiencies affecting cellular and humoral immunity (21.8%), congenital defects of phagocytes cells (18.7%), CID with associated or syndromic features (9.3%), defects in intrinsic and innate immunity (6.4%), and PIDs associated with diseases of immune dysregulation (3.2%). Complement deficiency was not detected in any of the patients studied.
Immunodeficiencies affecting cellular and humoral immunity were identified in patients with deficiency of T-CD3+ cells, T-CD4+ deficiency, T-CD8+ deficiency and CD40/CD40L deficiency. All showed normal levels of B and NK cells. One patient with T-CD3+ deficiency also showed a low level of IgG, suggesting a combined immunodeficiency. A major obstacle in diagnosing the pathogenesis of combined immunodeficiency is that different mutations in a single gene may lead to different clinical conditions and a similar clinical phenotype.25 A seven-year-old SCID patient had severe infections in the respiratory tract, gastrointestinal, skin, soft tissues, CNS, associated to lymphopenia, and a decrease in IgG and absence of proliferative response. We could not detect the defect in CD3ζ, CD3ɛ, p56lck and LAT (data not shown). In the case of the patient with CD8+ deficiency and normal CD4+ counts, a low molecular weight of Zap70 protein was observed. Zap70 is a tyrosine kinase, involved with TCR transduction signal together with the Lck and Fyn proteins. When there is an alteration in this protein, the TCR is not adequately formed and therefore T lymphocyte activation26 is impaired. The onset of disease occurred at seven months and was associated with severe gastrointestinal infections due to intracellular bacteria and parasites. The patient with CD4 lymphocyte deficiency died before molecular studies could be performed.
Driven by contact with T helper follicular cells, B cells undergo further diversification of their repertoire of antibodies in peripheral lymphoid organs. This occurs mainly through isotype-shift recombination events and somatic hypermutation. Binding of CD40 on the surface of B cells with CD40L expressed on T helper follicular27 leads to the transcription of target genes that result in the clonal expansion of B cells and the formation of the germinal center. The result of this overall process is the maturation and differentiation of the immunoglobulins and the maturation of antigen specificity. When this process fails, Hyper IgM Syndrome is produced.28 In our study, four cases were detected, one of them being a one-month-old male and the second a 14-month-old male patient. Two patients presented with very high IgM levels, with the other immunoglobulins at normal or reduced levels, and were associated with the absence of CD40 ligand (CD40L), a central element of the dependent T cell response. One patient was a seven-year-old female with an onset of symptoms from four months of age, presenting skin and respiratory infections with the isolation of extracellular bacteria and virus. The level of CD40 expression was demonstrated to be reduced. The fourth patient, a four-year-old male, died before the initial molecular study.
Predominantly antibody deficiencies have been described as the most frequent.29 These PIDs are associated with intrinsic defects, from the maturation of B lymphocytes in their different stages to alterations in the induction signals that affect the ability to generate and release the different types of immunoglobulins.30 In our patient group, one patient had a significant deficiency of B lymphocytes, accompanied by hypogammaglobulinemia. Several proteins have been implicated in the failure of B lymphocytes to develop normally. These include mutations in BTK, or the scaffold protein BLNK which account for approximately 90% of patients with defects in early B cell development.31 The expression of BTK and BLNK gene was measured by RT-PCR. A significant decrease in BLNK was observed, which is responsible for blockade of B-lymphocyte development and the agammaglobulinemia. Recently, four different monogenic defects leading to the CVID phenotype have been described, ICOS (inducible co-stimulator molecule), TACI (trans-membrane activator and calcium-Modulating cyclophilin ligand interactor-TNFRSF13B), BAFF-R (B cell activating factor-receptorTNFRSF13C) and CD19.32 In one CVID patient (10 years), the onset of disease was at seven years of age, and presented as respiratory infections and bronchiectasis. A deficiency in APRIL deficiency was demonstrated. None of the patients showed BAFF-R deficiency, CD19, or ICOS (data not shown). TACI was not studied in our group. The presence of bronchiectasis in this pathology is common, with a prevalence of 16–17%, and is usually diagnosed late since its clinical manifestations are variable and often go unnoticed.2 Although the genetic defects that lead to subclass defects have not yet been identified,8 we found three defects of IgG4 and one patient with IgG3. The clinical manifestation in these patients was mainly recurrent diarrhea and, in one of them, upper respiratory tract infections.
Chronic granulomatous disease (CGD) is a rare primary immunodeficiency disorder in which the microbicidal capacity of the phagocytic cells is reduced. This leads to recurrent and life-threatening bacterial and fungal infections in affected patients. CGD is caused by defects in one of the subunits of the phagocyte-specific components of nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase. The deficiency in p22-phox has been reported in 21% of AR-CGD.33 In Venezuela, we previously reported two CGD foci where the proteins involved were p47-phox and p67-phox.12,13 In four patients which presented with recurrent infections such as abscesses in the skin and lung, infection by Staphilococcus sp, in addition to Micobacterium sp and fungi, greatly reduced superoxide production, was observed. We were able to perform molecular analysis in three of these patients, and demonstrated the absence of p22-phox, suggesting a Chronic Granulomatous Disease. Deficits in Leukocyte Adhesion (LAD) were characterized in two newborn patients who presented with a late detachment of the umbilical cord, besides omphalitis and severe respiratory and skin infections associated with extracellular bacteria. The integrins β2 CD18/CD11 are proteins that participate in the adhesion between circulating leukocytes, endothelial cells and in the phagocytosis of complement coated particles, as part of the inflammatory response.34
Only one patient had a defect in IL-12 production, both in monocytes as in HLA-DR+ cells. Because he presented with BCGosis, Micoabterium tuberculosis infection, MSMD syndrome was suggested. The interleukin-12 (IL-12) of the macrophage is fundamental in the induction of the cellular type response. It has been reported that patients with MSMD who have genetic defects may show other infectious diseases, or even remain asymptomatic. Most of these inborn errors do not show complete clinical penetrance for the MSMD phenotype.35
Three patients with eczema, recurrent skin and lung infections, associated with Staphilococus aureus, viruses such as Cytomegalovirus and fungus such as Candida sp, had an elevation of serum IgE, 10 times above the reference value according to their age. All of them were identified as Hyper-IgE syndrome. This syndrome is associated with signaling impairment of the DOCK 8, STAT3, and TYK2 proteins, which affects coordination between Th17 effector cells, B cells and neutrophil response.36 In one patient, RT-PCR revealed a change in TYK2, the protein involved in the JAK-STAT signaling pathway (signal transducer and transcription activator), and in another, an alteration of the protein Dock8 and diminished of IL-17 production was found. This decrease in IL-17 may explain the susceptibility of this patient to recurrent candidiasis mucocutaneous, since it modulates neutrophil recruitment and stimulates the secretion of peptides such as beta defensins that play an important role in the skin's and lung defense mechanisms.23 The other patient is still under study, for the determination of their molecular alteration.
Lastly, we were able to detect a diminishment of XIAP expression associated with an autoimmune lymphoproliferative syndrome, an alteration of the apoptosis pathway that affects the overall survival of lymphocytes, leading to lymphoproliferation, autoimmune alterations and cancer.37 Our patient presented hepatosplenomegaly, which is a characteristic of lymphoid proliferation, which was associated with a severe EBV infection. In such patients, fluctuations in the size of the spleen and liver occur over time in 85% and 45% of cases.38 In general, lymphoproliferation improves with age. Numerous pathologies associated with programmed cell death involve the family of apoptosis inhibitory proteins.39
In this study we detected 17 types of PIDs in patients with recurrent infection. The most frequent major group of PID was humoral deficiencies followed by immune innate deficiencies. Knowing that these PIDs are present in our population and have been poorly diagnosed should provide an incentive to establish the appropriate laboratories to provide for more timely diagnosis and treatment, which could reduce the high costs involved, and lead to better quality of life for patients. Finally, the results presented in this study based on molecular, biochemical, subpopulation identification and functional assays allowed us to make the first approach to detecting defects in the immune response in patients with recurrent infections associated with PIDs; however, genetic sequence analyses are still to be done in order to identify the kind of mutation present in patients and their relatives that explain these defects.
Conflict of interestAll authors have no conflict of interest.
This work was supported by Grant CDCHTA-ULA M-1091-17-07-A.
