





The hepatitis B virus (HBV) surface antigen (HBsAg) variations suggested having some effects on infection outcome. Due to some controversial issues, the aim of this study was to compare the pattern of HBsAg variation between asymptomatic carriers and HCC/cirrhosis patients.
Materials and methodsIn this cross-sectional study, 19 HCC/cirrhotic and 26 asymptomatic patients were enrolled. After viral DNA extraction, HBs gene was amplified using an in-house nested-PCR. Then, PCR products were introduced into bi-directional Sanger sequencing. The retrieved sequences were compared with references, to investigate the variation of immunologic sites, major hydrophilic region (MHR) of HBsAg as well as reverse transcriptase (RT), and also to determine genotype/subtype.
ResultsThe analysis of MHR and epitopes on HBsAg showed dozens of substitution, which occurred more prevalently in I110, P120, Y134, G159, S193, Y206, S207, I208, L213 and P214 positions. However, Y134N/F/L (P=0.04) and P120T/S (P=0.009) were significantly detected in MHR and B-cell epitope of HCC/Cirrhotic group. A number of truncation-related mutations were higher in HCC/Cirrhotic group (P>0.001), albeit only C69* stop codon was statistically significant (P=0.003). In RT, some potentially resistant substitutions such as Q215S, V191I and V214A, were revealed. Phylogenetic analysis showed that all of isolates belonged to genotype D, and the major serotype was ayw1.
ConclusionThe higher frequency of substitutions in MHR and immune epitopes at positions such as Y134 and P120 as well as stop codons such as C69* in HCC/cirrhotic group might candidate them as predictive factors for infection outcome.
Even decades since the start of international coverage of hepatitis B virus (HBV) vaccine, HBV infection has still remained persistent in about 250 million individuals worldwide [1]. The chronic HBV infection remains asymptomatic or actively progresses toward cirrhosis and hepatocellular carcinoma [2]. The host factors including age, gender, race, and genetic background in addition to viral sequence diversity have been proposed to have ian mpact on the disease progression [3].
The DNA genome of HBV encompasses 4 open reading frames (ORF), which encodes 7 different proteins [4]. The virus polymerase, reverse transcriptase (RT) is an error-prone enzyme that leaves significant mutations alongside the viral genome during productive replication [5]. Interestingly, RT gene is overlapped by the small surface protein, Hepatitis B virus surface antigen (HBsAg) that constitutes as the major part of virus genome. As a result, mutations in the small HBsAg region interchangeably affect RT protein and vice versa [6].
HBsAg variants have been involved in some medical issues including failure of passive immunization by immunoglobulin (IG), disability of diagnostic method for detection, and immune evasion [7,8]. Variations that contribute to this issue are mainly located at the dominant epitope of HBsAg inside the “a” determinant (aa 124–147), within the major hydrophilic region (MHR, aa 99–169) [7].
The importance of HBsAg variations in immune evasion and disease progression has already been taken into consideration [7,9–13]. The detailed analysis of immunologic regions of HBsAg showed an association between some point mutations including substitution and deletion with immune escape phenotypes [7,14]. On the other hand, amino acid change in HBsAg structure could ultimately alter the virus secretion and replication [15]. While the point mutations that attributed to immune evasion have been defined clearly, those that are responsible for disease progression need to be investigated further. In fact, some substitutions such as T53C, L94S, S98T, P120T/S, A181T, L176, P203Q, S210R, and L213I are suggested to be involved in disease outcome [7,10,12]. Furthermore, the significance of some HBsAg stop mutations including L95*, Q104*, W172*, W182*, W196*, and L216* that are implicated in disease progression [12,16].
More precisely, some HBsAg mutations have been suggested to contribute to hepatocellular carcinoma (HCC)/cirrhosis development, although controversial issues remain to be elucidated. Regarding this issue, a genotype-dependent pattern of variation has been suggested inside the HBV genome [17,18]. For example, in some studies with the aim to find occult HBV-related mutations, the “a” determinant variations were significantly correlated with virus genotype [7,18]. Therefore, in order to justify this investigation, HBsAg mutations in disease progression should be conducted separately for each genotype.
On the basis of genome sequence, eight (A–H) different HBV genotypes were determined by Norder et al. [19]. HBV genotype D has been detected as the dominant genotype in Iran [20].
There is limited data regarding the comparison of HBsAg variations between HCC/cirrhotic cases and asymptomatic individuals amongst a pure population of patients with genotype D. Therefore, in this study the HBsAg mutation patterns between these groups were investigated and some new critical substitutions and stop codon mutations were suggested to be responsible for disease progression.
2Materials and methods2.1Patient's selectionIn this study, the subjects (n=46) were consecutively recruited from the Gastroentrohepatology and Liver Transplant Research Centers at Nemazee Hospital, Shiraz, Iran, from September 2013 to April 2016. All the cases were negative for antibodies against HCV, HIV and HDV viruses according to their medical records. None of them were vaccinated for HBV or had undergone IG therapy. Asymptomatic individuals (N=26) and HCC/cirrhotic cases (N=19) were primarily diagnosed and selected by a gastroenterologist based on their medical records including biochemical, virological, imaging and pathological examinations, which were also approved by a liver specialist. These patients did not receive any kind of antiviral drugs for at least 3 months prior to this study. Blood sample was collected in anticoagulant EDTA 5%, and serum was stored in −70 until further analysis. Prior to sampling, written informed consent was obtained from each participant. The study was approved by the local Ethics Committee of Shiraz University of Medical Sciences.
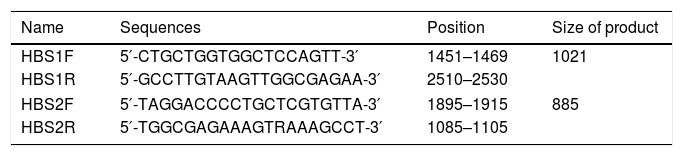
2.2Primer designThe amplification primers were designed regarding the validated genotype B, C and D sequences, using Primer Designing Tool from NCBI homepage and MEGA7™ software package (Table 1). They were selected not only to amplify the HBsAg region (aa 21–226), but also to cover critical drug-resistance mutations in the RT protein (aa 40–270) that overlap with the S region.
2.3Viral DNA extraction and HBs gene amplificationThe HBV DNA genome was isolated from 200μL of the serum sample, using a viral DNA extraction kit (Cinnagen Inc. Tehran, Iran), according to the manufacturers’ instruction. After DNA extraction, the samples were subjected to nested-PCR (polymerase chain reaction) amplification. The first-round of PCR was performed in 28 cycles of 94°C for 35s, 58°C for 45s and 72°C for 40s, and a final extension step at 72°C for 3min. The PCR reaction was prepared in a 25μL volume containing 5μL of extracted DNA, 200mM of each dNTP, 1.5mM MgCl2, 0.5pmol of each outer primer and 1U Taq DNA polymerase (Cinnagen Inc., Tehran, Iran). In the nested amplification step, 2μL of the first-round PCR product was subjected to PCR reaction containing HBSF2 and HBSR2 primers. To ensure the reliability of the tests, appropriate positive controls were also included.
2.4Sequencing and multiple sequences alignmentThe PCR products of the nested round were purified using PCR Product Purification Kit (MN Inc., Germany) and subjected to bi-directional traditional Sanger sequencing with inner primers (HBSF2 and HBSR2). The sequencing data were retrieved and aligned using MEGA7 software with different HBV genotype D sequences to find the mutations. All the differences of new sequences with HBV selected sequences were considered as mutations. A group of reference sequences, which was selected from HBV databank (https://hbvdb.ibcp.fr/HBVdb/HBVdbNomenclature) included as follow: X65259.1, AF121240.1, FJ904433.1, M32138.1 and, X85254.1.
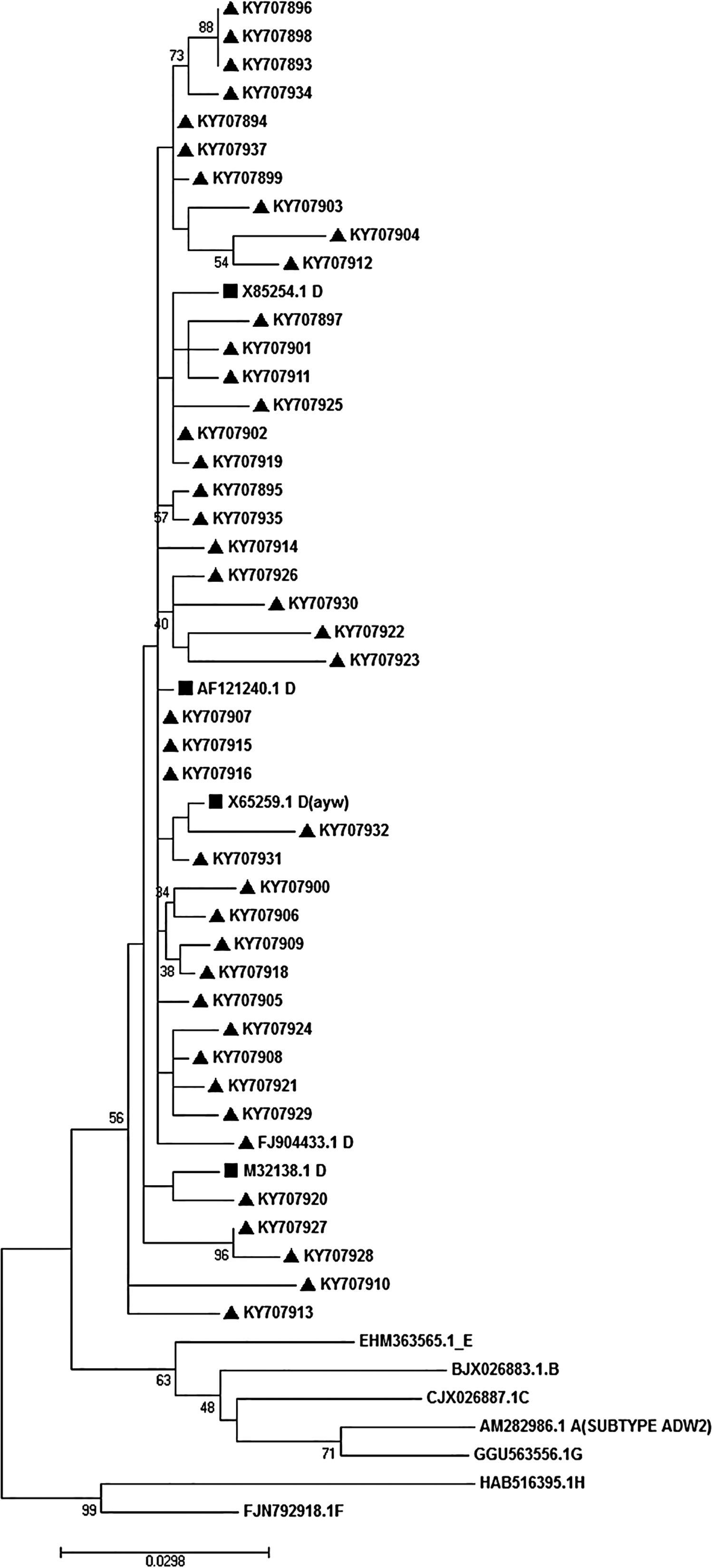
2.5Phylogenetic analysis and subtypingFor phylogenetic analysis, reference sequences of all HBV genotypes were entered into MEGA7 software and multiply aligned with new amplified sequences. The selected reference sequences were AB033559.1, X85254.1 and M32138.1 (genotype D); AM282986.1 (genotype A); AF282918.1 (genotype B); NC-003977.1 (genotype C); AB032431.1 and X75657.1 (genotype E) and AB275308.1 (genotype H). After alignment, phylogenetic analysis was performed following a maximum-likelihood method of tree construction. The Kimura-2 parameter substitution model was applied during the tree construction. To make sure, we also performed bootstrap resampling analysis using 200 replicates and presented them on the tree nodes. For subtyping, the relevant subtypes were determined regarding the algorithm, which was described by Yoshihiko Yano et al. [21,22]. In brief, considering the identity of amino acids at positions of 122 (Lys or Arg for d/y typing) and 160 (Lys or Arg for w/r typing) or 127 (Pro-Thr-Leu/Ile), the virus subtypes were determined.
2.6Statistical analysisStatistical analysis was performed in Epi Info™, using the Chi-square test; the significance level was determined<0.05.
3Results3.1Patient's dataIn the present study, 45 HBV patients including 26 asymptomatic and 19 HCC/cirrhotic cases were analyzed for their HBsAg sequence. Medical records including demographic and laboratory tests showed that the mean age of the participants in healthy carrier and HCC/cirrhotic patients were 39.2±15.1 and 50.5±12.6, respectively, which was statistically significant (P=0.031). Regarding participants’ gender, there were 34(75.6%) males and 11(24.4%) females. The ELISA assay also showed the presence of HBsAg in all the evaluated sera samples. The mean levels of ALT in asymptomatic and HCC/cirrhosis were 33±21 and 51±28 (P=0.018), respectively while for AST it was 28±8 in the asymptomatic and 97±82 in cirrhotic/HCC groups (P=0.0001).
3.2Analysis of important immunologic regions of HBsAg sequenceThe comparison of immunologic regions of all cases revealed not only some pre-defined variations with expected immune consequences, but also dozens of novel mutations with unknown consequences. The list of all variations of the immunologic site is presented in Table 2. In total, the sequence analysis detected more than 160 amino acid substitutions in all the samples. Out of which, 58.4% occurred in the HCC/cirrhotic group and 41.6% in the asymptomatic cases (P>0.05). The epitope regions responsible for B-cell and T-cell immunity are shown in Table 2. According to their position, our results revealed 51, 36 and 45 amino acid substitutions in B-cell, Th (CD4) and CTL epitopes, respectively. These findings showed a high prevalence of amino acid substitutions of Y134H/F/L (N=8), P120S/T (N=5), I110L (N=5) and G159V/A/*(N=4) inside the B-cell epitope site. The substitutions of Y134H/F/L (P=0.04) and P120T/S (P=0.009) in this epitope showed to be more prevalent in the HCC/cirrhotic group. In addition, S193N/L (N=5), C221Y (N=3) and I82T (N=3) were detected as more prevalent substitutions inside T-helper epitope site. For CTL epitopes, substitutions including Y206F/L/S/H/C (N=8), S207R (N=7), I208T (N=5) and P214L/Q (N=5) were detected more frequently than other mutations in the two groups.
The immune evasion variations regarding the major B or T cell epitope sites. The variations are ordered by locations and cell subsets.
| Amino-Acid Substitutions/Mutations of HBsAg | Cell subsets | Sequence |
|---|---|---|
| Q101R(3), M103T(1), L104W(1), V106D(1), C107*(1), L109P(1), I110L/T(5), S114P(1), T115N(1), T118A(1), P120S/T(5), R122T(1), C124G(1), T126R(3), P127T(1), G130K/R(3), M133I(1), Y134H/F/L(8), T140I(2), K141E(1), D144A(1), T148I(1), I152S(1), A157V(1), F158V(1), G159V/A/*(4) | B cell | 100–160 |
| – | Th (CD4) | 19–28 |
| Q30K(1), G44E(1), L49R(1), S53L(1), Q54R(1), P62L/Q(2), S64F(1) | Th (CD4) | 21–65 |
| F80S(1), I82T(3), F85C(1), L88P(2), L89Q(1), I92T(1), L94*(1), L95*(1), V96A(1) | Th (CD4) | 80–98 |
| L186F/C(2), T189I(1), S193L(5), M197T(1) | Th (CD4) | 186–197 |
| L215P(1), L216*(1), P217L(1), F220S(1), C221Y(3) | Th (CD4) | 215–223 |
| V177L/A(2), F179S(2), Q181H(1), W182L(1)/*(2), V184G(2) | CTL (CD8) | 171–184 |
| Y206F(2)/L(1)/S(3)/H(1)/C(1), S207R(3)/N(1)/T(3), I208T(5), L209S(1), S210K(1)/T(1)/R(1), F212S(1), L213I(4), P214L(4)/Q(1), L215P(1) | CTL (CD8) | 206–215 |
Regarding the overlaps of MHR and B-cell epitope, they exhibited a similar pattern of variation. The important MHR substitutions with defined functional effects are listed in Table 3. These variations that are accounted for vaccine or IG immunotherapy escapes were found in 8.3% and 16.7% of all cases. The sequence analysis showed that important substitutions including P120S/T (N=5), R122T (N=1) and T140I/N (N=3, P>0.05) were detectable. Interestingly, no sign of critical G145R mutation was probed in all the cases. Furthermore, some other mutations such as S114P, T115N, T118A, T124G, T126R, P127T, G130/*/K/R, M133R, Y134H/F/L, K141E, D144A, T148I and I152S were also detected amongst the MHR region with less effective consequences.
The substitutions alongside the MHR region.
| Substitution in MHR | Number of cases | Vaccine escape | Immunotherapy escape | Failure of HBsAg detection |
|---|---|---|---|---|
| P120S | n=2 | + | − | + |
| P120T | n=3 | − | + | + |
| R122T/K | n=1 | + | − | + |
| M133I | n=0 | − | + | + |
| T140I/N | n=3 | − | + | − |
| S143L | n=0 | − | − | + |
| G145R | n=0 | + | + | + |
| Substitutions with unknown functions | S114P(1), T115N(1), T118A(1), C124G(1), T126R(1), P127T(1), G130K(2), G130R(1), G130*(1), M133R(1), Y134N/F/L(8), K141E(1), D144A(1), T148I(1), I152S(1) |
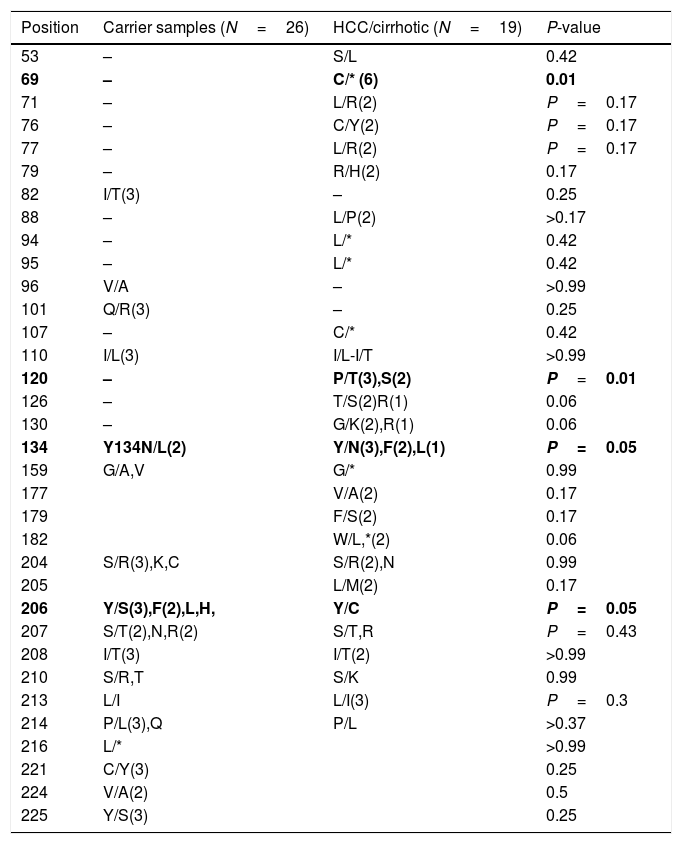
After multiple alignments of all retrieved sequences, mutation patterns for the samples were revealed. As a result, the comparison of variations between HCC/cirrhosis and asymptomatic carrier samples also showed the differences between groups, as listed in Table 4. Substitutions of P120T/S were significantly higher in the HCC/cirrhotic patients (5/19 cases, 26%) than those with asymptomatic condition (0/26, 0%) (P=0.009). Moreover, Y134N/F/L was seen more frequently in the HCC/cirrhotic patients (6/19 cases, 32.2%) compared to those with asymptomatic condition (1/26, 3.9%) (P=0.04). Furthermore, a stop codon mutation of C69*(6 cases=32.2%) was detected more in the HCC/cirrhotic patients (P=0.003).
The comparisons of variations among HCC/cirrhosis with asymptomatic carrier samples.
| Position | Carrier samples (N=26) | HCC/cirrhotic (N=19) | P-value |
|---|---|---|---|
| 53 | – | S/L | 0.42 |
| 69 | – | C/* (6) | 0.01 |
| 71 | – | L/R(2) | P=0.17 |
| 76 | – | C/Y(2) | P=0.17 |
| 77 | – | L/R(2) | P=0.17 |
| 79 | – | R/H(2) | 0.17 |
| 82 | I/T(3) | – | 0.25 |
| 88 | – | L/P(2) | >0.17 |
| 94 | – | L/* | 0.42 |
| 95 | – | L/* | 0.42 |
| 96 | V/A | – | >0.99 |
| 101 | Q/R(3) | – | 0.25 |
| 107 | – | C/* | 0.42 |
| 110 | I/L(3) | I/L-I/T | >0.99 |
| 120 | – | P/T(3),S(2) | P=0.01 |
| 126 | – | T/S(2)R(1) | 0.06 |
| 130 | – | G/K(2),R(1) | 0.06 |
| 134 | Y134N/L(2) | Y/N(3),F(2),L(1) | P=0.05 |
| 159 | G/A,V | G/* | 0.99 |
| 177 | V/A(2) | 0.17 | |
| 179 | F/S(2) | 0.17 | |
| 182 | W/L,*(2) | 0.06 | |
| 204 | S/R(3),K,C | S/R(2),N | 0.99 |
| 205 | L/M(2) | 0.17 | |
| 206 | Y/S(3),F(2),L,H, | Y/C | P=0.05 |
| 207 | S/T(2),N,R(2) | S/T,R | P=0.43 |
| 208 | I/T(3) | I/T(2) | >0.99 |
| 210 | S/R,T | S/K | 0.99 |
| 213 | L/I | L/I(3) | P=0.3 |
| 214 | P/L(3),Q | P/L | >0.37 |
| 216 | L/* | >0.99 | |
| 221 | C/Y(3) | 0.25 | |
| 224 | V/A(2) | 0.5 | |
| 225 | Y/S(3) | 0.25 |
A substitution of Y206S/F/L/H/C was frequently detected (7 vs. 1 cases) in asymptomatic cases in comparison with the HCC/Cirrhotic patients (P=0.06). Although the frequency of T126S/R, G130K/R and W182L/* mutations were detected in 3 out of 19 of the HCC/cirrhotic group and none of the asymptomatic cases, they were not statistically significant (P>0.05). Also, I82T (N=3), Q101R (N=3), S207T/N/R (N=5), P214L/Q (N=4), C221Y (N=3) and Y225S (N=3) mutation was detected more prevalently in the asymptomatic group than those with HCC/cirrhosis, (P>0.05).
Stop codon mutations which cause a truncated form to HBsAg including C69*(N=6), W182*(N=2), L94*, L95*, C107* and G159* were observed in 47.4% of individuals in the HCC/cirrhotic group, but only one (L216*) was detected among the asymptomatic cases (P>0.001). However, among the truncation-related stop codons, only C69* was significantly different between the two studied groups (P<0.003).
3.4Drug resistance pattern in polymerase geneAs the S gene sequence overlaps polymerase region, some substitutions in S gene might leave a profound effect on RT drug susceptibility. This study showed that a part of substitutions along with S gene leads to missense variations in RT coding sequence, simultaneously. As a whole, 13 substitutions with medical importance were detected inside the RT region. The analysis revealed that just one primary substitution, A194T, responsible for complete drug resistance to Adefovir or Tenofovir was detected. The most prevalent mutation was Q215S (N=6), which is a potential resistance mutation, partially destroying RT drug responsiveness to adefovir and lamivudine. Some other variations with less medical importance including V191I (N=2), I169T (N=1), V214A (N=2) and I233V (N=1) were detected in a lower frequency.
3.5Determination of HBV genotype, subgenotype, and serotypesThe HBs sequences of all 45 isolates (the temporal accession numbers: KY707893–KY707937) were compared with the reference sequences of 8 HBV defined genotypes. The Maximum-Likelihood phylogenetic tree construction method was employed for genotype determination. The phylogenetic analysis showed that all the analyzed sequences belonged to genotype D, as depicted in Fig. 1. The survey of the serotype also revealed that the majority of samples (42 out of 45) were ayw1 serotype, while the rest were determined as ayw3 and ayw2, respectively.
4Discussion
HBsAg variants have been suggested to be involved in some issues, such as vaccine escape, failure of IG protection, a shortcoming in the detection of carriers, and even in disease progression (10). While the correlation of some HBsAg variations with disease progression has been suspected (10), it still remains to be clarified further.
As the main aim of this study, the comparison of the variation pattern between groups showed that some substitutions such as of Y134N/F/L and P120T/S were significantly more prevalent in the HCC/Cirrhotic cases than the asymptomatic group. In a similar finding, P120 position was shown to be a critical point for HCC/cirrhosis prediction [4]. However, Y134N/F/L has not been reported as an important position for HCC/cirrhosis prediction. Instead, a correlation between this mutation and HBV reactivation following the immune suppression was reported [4,21,22]. In spite of immune evasion mutations, the controversial issue still remain unclear for the role of HBsAg variations in disease progression. The studies that were in agreement with the role of HBsAg variation in disease progression suggested a genotype-dependent manner [17,18]. It was shown that occult HBV related substitutions strongly follow a genotype-dependent pattern [17,18]. Mutations in T53, L94, S98, P120, A181, L176, P203, S210, and L213 positions have been suggested to be involved in the disease outcome [7,10,12]. However, none of them was detected as a remarkable variation in our experiment.
As an interesting finding, a noticeable number of stop codons were detected among the HCC/cirrhotic group. In total, 12 stop codon mutations including C69*, C107*, G159*, W182*, L94* and L95* were detectable, but only C69* was significantly important. Regarding the length of HBsAg, it is predictable that truncation from gene end such as L216 leaves an ineffective alteration in the structure when compared to longer deletions of protein (e.g. C69 and C107) in the HCC/Cirrhotic group. This is not the first report claiming the importance of HBs truncation in disease progression. These long truncations lead to deletion of carboxyl terminal and creation of new HBsAg versions with different activities. Interestingly, stop codons including L95*, Q104*, W172*, W182*, W196* and L216* have been indicated to be responsible for disease progression toward tumor and cirrhosis [12,16,23]. The stop codon mutations like L95*, W182*, and L216* were shown to activate cell proliferation and induce transformation through transactivation of different oncogenes [16,23]. Similarly, the presence of stop codons in our HCC/cirrhotic group might also emphasize on their molecular roles in enhancing the disease progression toward cirrhosis and HCC by unknown mechanisms. The correlation of C69* stop mutation with cirrhosis has also been described by Veazjalali et al. on a pure genotype D infected population [24]. However, since there is no similar report, a genotype dependency for these kinds of mutations is predictable.
In addition, truncating stop codons are suspected to correlate with failure in HBsAg detection and with virus diagnosis [11,25]. On the contrary, all of our cases showed a detectable amount of HBsAg when assayed by an ELISA method. Hence, it could be concluded that at least in our population with genotype D, the role of variations in detection failure is ignorable. By modeling and in vitro assessment, Sadeghi et al. showed that variations in MHR might have an impact on diagnosis, but not as much to entirely destroy the detection process [15].
In the MHR region, substitutions of Y134N/F/L and P120T/S were detected more in the HCC/Cirrhotic cases compared to the asymptomatic carriers. Several mutations in the MHR region and HBsAg have been described, which leads to the emergence of immune escapes mutant [8,12,27]. The more frequent immune evasion mutations reported so far include P120T, R122K/T, Q129H, M133I, F/Y134N, S143L, D144E/A, G145R, E164D, A168V, W172*, W182*, and I195M [6,26–28]. Amongst these mutations, Y134 and P120 positions showed a higher prevalence of variations in our study. All these mutations cause the virus to escape from vaccine, IG, and commercial detection assays [26]. In our study, the well-known G145R substitution, as a remarkable vaccine escape mutation, was not detected. In line with our study, G145R has been reported in a very low frequency (<0.5%) in other studies [6,9,26,27]. It is suggested that mutations including G119R, C124Y, I126S, Q129R, S136P, C139R, T140I, K141E, D144A, and G145R significantly impaired the virus and S protein secretion in vitro[7,11,15]. Therefore, mutations in the MHR or in other immunological regions might also impact the virus replication, and consequently the disease outcome.
In the case of cellular immune epitopes, our analysis revealed that S193 variation at T-helper epitope as well as some substitutions including Y206F/L/S/H/C, S207R, I208T, P214L/Q in T-cytotoxic epitopes were more prevalent. These findings also showed that substitution of Y206S/F/L/H in the CTL epitopes was detected more frequently in our study population. Some mutations in HBsAg immunologic region, like G159 and L213, were also detected with a lower frequency. In a similar report on genotype D samples, substitution at S207, I208 and L213 was determined as the most prevalent immune escape mutations [29]. The limit in HBsAg detection, cellular immune evasion and consequent viral persistence might be associated with both Th or CTL epitopes variations [31].
The substitution at position Y206 was detected more frequently, but was not significant in asymptomatic patients in comparison with the HCC/cirrhosis group. Its correlation with disease outcome was poorly described; however, the association with lower levels of virus titer has already been suggested. Mirabelli et al. suggested that specific HBsAg-mutations (M197T, S204N, and Y206C/H) were significantly associated with lower (<2000IUml−1) viral titer in the sera [30].
The analysis of RT drug resistance showed a low frequency of resistance-related substitutions. In all cases, just one primary resistance mutation (A194T) was confirmed, while other mutations such as Q215S/H were defined as secondary (potential resistance) [8,31]. It has been reported that the genotype D of HBV is associated with an increased risk of adefovir resistance. The position of Q215 from RT has been detected as more frequent spontaneous mutation arising among genotype D, and its mutants are potentially resistant to adefovir[32]. The low frequency of all RT mutations can be explained by the drug avoidance of the selected groups, which was reported in several studies [27,32].
In agreement with our results, several studies from the southern region of Iran have implied the dominant prevalence of genotype D throughout the country [20,29]. However, our study showed that serotype ayw1 was dominant amongst the investigated population. Another report indicated that serotypes ayw2 (94.4%) followed by ayw1 and ayw3 were more frequent than other types in our region [33].
Generally, this study highlighted the significant prevalence of some mutations inside the HBsAg in terms of their possible effects on disease progression; however, our findings should be evaluated in a larger population to meet the predictive marker criteria. Hence, participants must be age-matched to prevent bias with respect to the effect of disease duration. Moreover, traditional sequencing does not have enough sensitivity to detect mutations that are the minority. Therefore, in order to precisely define medically important mutations, performing clonal sequencing or next-generation ones could be the preferred choice.
In conclusion, the higher frequency of substitutions in MHR and immune epitopes at positions such as Y134 and P120 as well as stop codons such as C69* might have the predictive value to determine the HBV infection prognosis due to higher numbers in the HCC/cirrhotic group.AbbreviationsHBV hepatitis B virus hepatitis B surface antigen open reading frame reverse transcriptase immunoglobulin polymerase chain reaction
Study concept: Sarvari J, Malek-Hosseini SA, Fattahi MR; Study design: Sarvari J, Hosseini SY and Sanaei N; Bench work: Sanaei N; Patients selection: Fattahi MR and Malek-Hosseini SA; Data analysis: Sanaei N, Hosseini SY and Sarvari J; Manuscript drafting: Hosseini SY, Sarvari J; Critical revision of the manuscript: Fattahi MR and Malek-Hossini SA.
Financial supportThe present study was extracted from the thesis written by Neda Sanaei, which was financially supported by Shiraz University of Medical Sciences (Grant No. 94-10841).
It is declared that informed patients consent was obtained for publication.
Conflict of interestAll the authors declare no conflict of interest.
The present study was conducted by the help of Liver Clinic staff of Shahid Motahari Clinic complex who prepared patient samples. The authors wish to thank Mr. H. Argasi at the Research Consultation Center (RCC) of Shiraz University of Medical Sciences for his invaluable assistance in editing this manuscript.



