



Cirrhotic patients with hepatitis C virus genotype 3 infection show unsatisfactory outcomes after 12 weeks’ treatment with direct antiviral agents. The National Italian Drug Agency allows 24 weeks of therapy in difficult-to-treat patients, including genotype 3 cirrhotics. Aim of this study was to evaluate efficacy and safety of a 24-week course of sofosbuvir plus daclatasvir±ribavirin in this population.
Materials and methods106 consecutive cirrhotics (70.8% males, mean age 55.3±7.6 years) in 8 tertiary hepatology centers received sofosbuvir plus daclatasvir for 24 weeks. Ribavirin was administered in 85 (80.2%) based expected tolerability, at a mean dose of 964±202mg/day. Baseline Child-Pugh class was A 91.5%, B 6.6%, C 1.9%; mean baseline MELD was 8.5±2.7.
ResultsAll patients completed 12-week follow-up post-treatment, and 104 (98.1%) obtained sustained virological response (100% in ribavirin -treated patients vs. 90.4% without ribavirin; p=0.04). No worsening in renal and liver function was observed, no serious adverse events occurred. Two virological failures showed resistance associated variants (Y93H and S282T).
ConclusionAn extended 24-week treatment with sofosbuvir plus daclatasvir+ribavirin obtained 100% efficacy in genotype 3 hepatitis C cirrhosis, with very limited side effects. The role of ribavirin seems crucial in this setting and should be administered if clinically feasible.
Chronic hepatitis C virus (HCV) infection is the main cause of chronic liver disease in Italy, where although the exact number of infected subjects is unknown, a prevalence of approximately 1% is estimated in the general population [1]. By February 2017 roughly 70,000 infected patients had been treated with new direct antiviral agents (DAA): initially only patients with advanced liver disease, transplanted recipients or with extrahepatic manifestations were eligible to receive DAAs. Of these, approximately 43,450 cirrhotics were treated with an overall sustained virological response (SVR) higher than 90% [1]. However even with these new regimens cirrhotic patients, especially if infected by HCV genotype (G) 3, remain difficult to treat. HCV G3 is the second most prevalent genotype worldwide with an estimated prevalence of 30% among all HCV infected subjects in Europe [2,3]. G3 is associated with a higher risk of steatosis and hepatocellular carcinoma (HCC) [4]. The first interferon-free combination based on sofosbuvir (SOF) plus ribavirin (RBV) for 24 weeks reached suboptimal SVR rates in cirrhotic patients [5,6], leading to a search for other alternative combination regimens. The combination of SOF plus daclatasvir (DCV) for 24 weeks instead of 12 was recommended based on expert opinion [7], as no data from randomized clinical trials in cirrhotic patients have been published in the Italian patient population.
The ALLY-3 trial demonstrated a very low SVR rate (approximately 60%) among G3 cirrhotic patients treated with SOF+DCV without RBV for 12 weeks [8]; the extension of treatment duration to 16 weeks, and the addition of RBV in the only randomized study ALLY-3+ increased SVR to 86% [9]. Three Italian real-life studies published in abstract form [10–12] reported an increase of SVR to 90–98% with 24 weeks’ duration and some benefit from RBV administration. Also data produced by the UK EAP confirmed how RBV is significantly associated with SVR12 in G3 cirrhotic patients [13].
Given the lack of clear-cut data regarding the ideal antiviral regimen in HCV G3 cirrhotic patients, the present study aims to investigate the efficacy and safety of a 24 week course of SOF plus DCV with or without RBV in this difficult-to-treat population.
2MethodsThis was a prospective multicenter study conducted in 8 tertiary hepatology centers in Lazio region, Italy from June 2015 to October 2016.
Eligibility criteria were: adult patients (age≥18 years) with HCV G3 infection and clinical evidence of cirrhosis. Diagnosis of cirrhosis was made based on liver biopsy or non-invasive fibrosis testing (liver stiffness measurement through transient elastography). Cirrhosis was defined histologically by a METAVIR score of F4 on biopsy or non-invasively by a liver stiffness >14kPa. Patients with decompensated cirrhosis were also included.
Treatment experienced patients were allowed with the exception of prior use of NS5A inhibitors. Patients who had been treated with SOF+RBV were included except for those who discontinued therapy for intolerance.
Exclusion criteria were: previous treatment with NS5A inhibitors, HCC beyond Milan criteria, renal insufficiency (glomerular filtration rate <30ml/min).
All patients received SOF 400mg plus DCV 60mg orally daily for 24 weeks as authorized by the Italian Medical Agency (AIFA). RBV was prescribed at physicians’ discretion, based on clinical features (i.e., previous decompensation, baseline hemoglobin levels) and/or expected tolerability (i.e., previous RBV therapy). The study was conducted in accord with the principles of good clinical practice and the declaration of Helsinki, and all patients provided written informed consent.
Genotyping was performed using the real time HCV genotype II assay (Abbott Molecular, Des Plaines IL, USA); quantitative HCV RNA levels were assessed at baseline, at treatment weeks 4, 12, 24 and at post treatment weeks 4, 12 using the HCV Abbott real time polymerase chain reaction (PCR) with a lower limit of quantification of 12IU/ml.
SVR12 was defined as undetectable HCV RNA at 12 weeks after treatment completion. Safety and tolerability were recorded at every visit. Resistance testing for variants (RAVs) in both NS5A and NS5B regions were performed by sequencing plasma HCV RNA from any patient who experienced virological relapse during and post-treatment.
2.1Statistical analysisA descriptive analysis was performed for socio-demographic parameters, as well as clinical variables related to liver function at baseline. Mean, standard deviation, range, median, interquartile range, number of missing data, and 95% confidence intervals were determined for continuous variables; absolute frequency, 95% confidence intervals and number of missing data for categorical variables. Associations between qualitative variables were measured with a χ2 test, while differences between quantitative variables were evaluated with a Student's t test. Statistical computations were performed with the SPSS® Statistics package version 20.0.0. Data were analyzed according to intention to treat (ITT).
3Results106 G3 cirrhotic patients were enrolled (70.8% males, mean age 55.3±7.6 years).
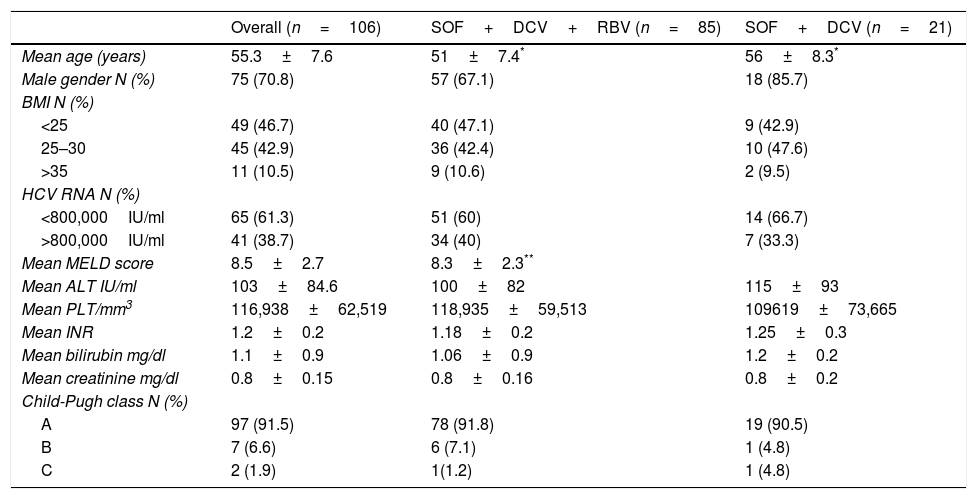
Baseline Child-Pugh class was as follows: A 91.5%, B 6.6%, C 1.9%. Baseline characteristics are listed in Table 1.
Baseline patients’ characteristics of cirrhotic patients infected with hepatitis C virus genotype 3 treated with sofosbuvir plus daclatasvir with or without ribavirin for 24 weeks.
| Overall (n=106) | SOF+DCV+RBV (n=85) | SOF+DCV (n=21) | |
|---|---|---|---|
| Mean age (years) | 55.3±7.6 | 51±7.4* | 56±8.3* |
| Male gender N (%) | 75 (70.8) | 57 (67.1) | 18 (85.7) |
| BMI N (%) | |||
| <25 | 49 (46.7) | 40 (47.1) | 9 (42.9) |
| 25–30 | 45 (42.9) | 36 (42.4) | 10 (47.6) |
| >35 | 11 (10.5) | 9 (10.6) | 2 (9.5) |
| HCV RNA N (%) | |||
| <800,000IU/ml | 65 (61.3) | 51 (60) | 14 (66.7) |
| >800,000IU/ml | 41 (38.7) | 34 (40) | 7 (33.3) |
| Mean MELD score | 8.5±2.7 | 8.3±2.3** | |
| Mean ALT IU/ml | 103±84.6 | 100±82 | 115±93 |
| Mean PLT/mm3 | 116,938±62,519 | 118,935±59,513 | 109619±73,665 |
| Mean INR | 1.2±0.2 | 1.18±0.2 | 1.25±0.3 |
| Mean bilirubin mg/dl | 1.1±0.9 | 1.06±0.9 | 1.2±0.2 |
| Mean creatinine mg/dl | 0.8±0.15 | 0.8±0.16 | 0.8±0.2 |
| Child-Pugh class N (%) | |||
| A | 97 (91.5) | 78 (91.8) | 19 (90.5) |
| B | 7 (6.6) | 6 (7.1) | 1 (4.8) |
| C | 2 (1.9) | 1(1.2) | 1 (4.8) |
SOF, sofosbuvir; DCV, daclatasvir; RBV, ribavirin; BMI, body mass index; MELD, model for end-stage liver disease; ALT, alanine aminotransferase; PLT, platelet count; INR, international normalized ratio.
According to previous antiviral treatment 61% patients were naïve, 39% were treatment-experienced (22 relapsers and 19 non-responders to peginterferon plus ribavirin). Only 1 patient had previously received SOF.
Eighty-five patients received RBV (80.2%) while 21 received only SOF+DCV (19.8%).
Mean RBV dose was 964±202mg/daily.
Baseline characteristics between the two treatment groups (with or without RBV) showed a significantly younger age and a lower baseline MELD among RBV-treated patients (p=0.008 and 0.017, respectively; Table 1). Nine patients with decompensated cirrhosis (Child-Pugh class B and C at baseline) all had ascites; 27 patients (25.5%) had esophageal varices at baseline. Mean pre-treatment liver stiffness measurement was 23.2±9.4kPa.
All patients completed 24 weeks of treatment with SOF+DCV±RBV. No patient was lost to follow-up and safety data was recorded at every scheduled or on-demand visit.
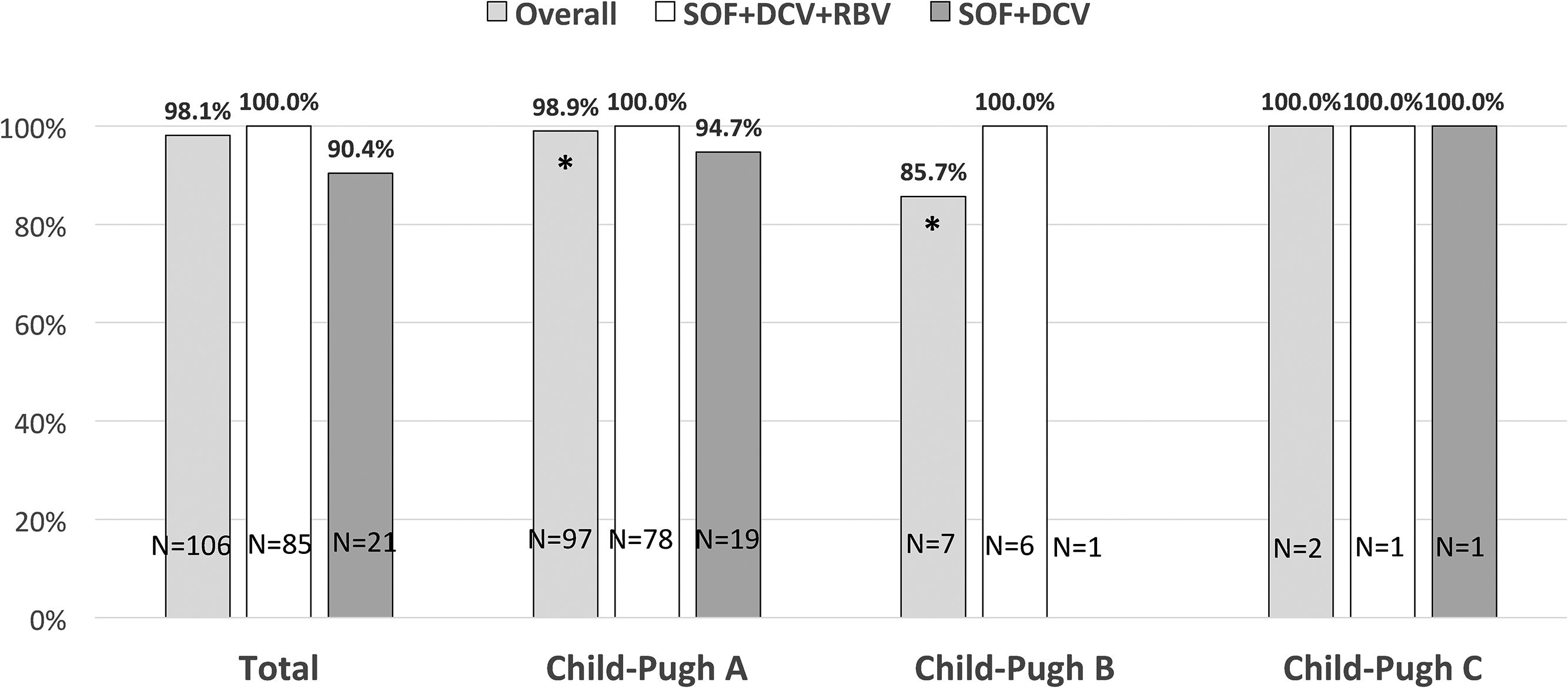
3.1Virological resultsAll patients were HCV RNA negative at end of treatment and SVR was obtained in 104/106 patients overall (98.1%) while SVR was 100% and 90.4% in the SOF+DCV+RBV and SOF+DCV group, respectively (p=0.04).
SVR based on Child-Pugh class at baseline showed a statistically significant difference for class A patients vs. class B patients (98.9% vs. 85.7% respectively, p=0.01) (Fig. 1).
 rates in cirrhotic patients infected with hepatitis C virus genotype 3 treated with sofosbuvir plus daclatasvir with or without ribavirin according to Child-Pugh class at baseline. Patients in Child-Pugh class A overall had significantly higher SVR rated compared to Child-Pugh class B patients (*p=0.01). SOF, sofosbuvir; DCV, daclatasvir; RBV, ribavirin.")
Sustained virological response (SVR) rates in cirrhotic patients infected with hepatitis C virus genotype 3 treated with sofosbuvir plus daclatasvir with or without ribavirin according to Child-Pugh class at baseline. Patients in Child-Pugh class A overall had significantly higher SVR rated compared to Child-Pugh class B patients (*p=0.01).
SOF, sofosbuvir; DCV, daclatasvir; RBV, ribavirin.
Two patients relapsed during follow-up; both received SOF+DCV and had obtained serum HCV RNA undetectability at week 4 of therapy. The first patient was non-responder to a previous IFN-based therapy, with good residual liver function (Child-Pugh class A, MELD 8) and 5 log serum HCV RNA at baseline. The second relapser was naïve, with decompensated liver disease (Child-Pugh class B, MELD 15) refractory ascites and 4 log serum HCV RNA at baseline. The analysis of RAVs at post treatment week 12, showed the development of Y93H in both patients and S282T in the latter only.
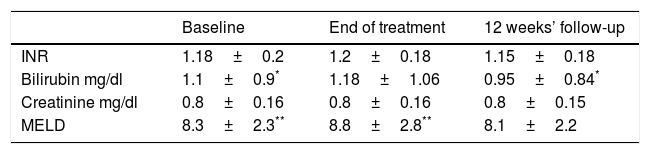
3.2Liver and kidney functionOverall no worsening in liver and kidney function was observed between baseline and end of follow-up; a increase in MELD score was found at end of treatment vs. baseline in patients who received ribavirin (Table 2), and returned to baseline levels at end of follow-up.
Mean liver and kidney function variables during treatment and follow-up in 85 cirrhotic patients treated with sofosbuvir plus daclatasvir plus ribavirin for 24 weeks.
| Baseline | End of treatment | 12 weeks’ follow-up | |
|---|---|---|---|
| INR | 1.18±0.2 | 1.2±0.18 | 1.15±0.18 |
| Bilirubin mg/dl | 1.1±0.9* | 1.18±1.06 | 0.95±0.84* |
| Creatinine mg/dl | 0.8±0.16 | 0.8±0.16 | 0.8±0.15 |
| MELD | 8.3±2.3** | 8.8±2.8** | 8.1±2.2 |
SOF, sofosbuvir; DCV, daclatasvir; RBV, ribavirin; INR, international normalized ratio; MELD, model for end-stage liver disease.
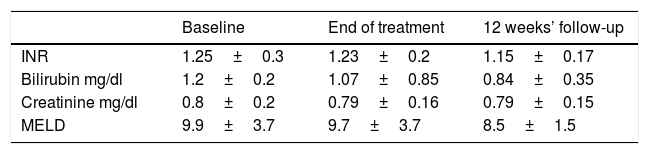
Mean liver and kidney function variables during treatment and follow-up in 21 cirrhotic patients treated with sofosbuvir plus daclatasvir for 24 weeks.
| Baseline | End of treatment | 12 weeks’ follow-up | |
|---|---|---|---|
| INR | 1.25±0.3 | 1.23±0.2 | 1.15±0.17 |
| Bilirubin mg/dl | 1.2±0.2 | 1.07±0.85 | 0.84±0.35 |
| Creatinine mg/dl | 0.8±0.2 | 0.79±0.16 | 0.79±0.15 |
| MELD | 9.9±3.7 | 9.7±3.7 | 8.5±1.5 |
SOF, sofosbuvir; DCV, daclatasvir; INR, international normalized ratio, MELD, model for end-stage liver disease.
No significant variations in INR, serum bilirubin, creatinine level were seen between baseline and end of follow-up within each of the 2 treatment groups (Table 2).
No significant variations in INR, serum bilirubin, creatinine level and MELD were seen between the 2 treatment groups.
No compensated patient experienced decompensation during treatment and follow-up. Two patients with decompensated cirrhosis (baseline Child-Pugh class C) remained stable during treatment and follow-up with no worsening in MELD score.
3.3SafetySOF+DCV±RBV combination was well tolerated. All patients completed treatment without any early discontinuations due to adverse events (AEs).
The most common reported AE was grade 1–2 anemia in 30 patients in the SOF+DCV+RBV group (35.3%), requiring RBV dose reduction in 10 patients (11.7%).
Overall 8 patients reported headache and irritability (7.5%; 2 in SOF+DCV group and 6 patients in SOF+DCV+RBV group, respectively).
Total AEs were significantly more common in the SOF+DCV+RBV group (42.3% vs. 9.5%, p<0.01).
No serious AEs were observed during treatment and follow-up.
4DiscussionThis real-life clinical study demonstrates an excellent safety and efficacy of SOF+DCV combination in a difficult-to-treat population of cirrhotic patients infected by HCV G3.
The extension of therapy to 24 weeks with the addition of RBV, both proposed by Italian AIFA rules and empirically suggested by EASL guidelines, allowed to achieve a higher SVR rate compared to a published randomized controlled trial with a treatment duration of 16 weeks. In fact in the present study G3 cirrhotic patients obtained an overall SVR rate of 98.1% compared to 86% reported in the ALLY-3+ trial [9]. This virological improvement appears to be even more significant if one compares data from RBV-treated patients in both studies: 100% in the present work vs. 86% in ALLY-3+. Therefore our data underscores the importance of prolonging treatment duration to 24 weeks, as well as adding RBV when feasible, to obtain an improvement in SVR. These findings are supported by and consistent with other three reports in abstract form from other Italian regions [10–12] and two European studies [13,14]. The beneficial role of RBV in treating G3 cirrhotic patients is in fact suggested by improved virological rates in RBV-containing regimens also in the UK EAP study though the total number of patients treated without RBV was small and robust conclusion cannot be drawn [13]. Similar results are reported in the Scandinavian study where the addition of RBV even for 12 weeks’ treatment showed a trend toward higher SVR12 [14]. However, our results are in contrast with a sub-analysis of an heterogeneous French early access program study by Hezode et al., which showed a lower SVR rates in 205 G3 cirrhotic patients treated for 24 weeks with the same regimens: SOF+DCV had 86% SVR12, while SOF+DCV+RBV had 82% SVR12 [15]. Therefore, in the study by Hezode et al., neither the extension of duration to 24 weeks, nor the addition of RBV appeared to improve SVR rates. A possible explanation for this difference in SVR, could be the characteristics of the French cirrhotic population which showed a higher rate of decompensated cirrhosis (15% vs. 8.5% in the present study) and a baseline MELD >15 in 30% of patients. Both these factors are known to negatively impact prognosis for virological success. Furthermore, baseline liver function was more severely impaired in the French population, accounting for the higher number of serious AEs in the group treated with RBV. All reported deaths and serious AEs were, in fact, observed only in patients with a baseline MELD ≥15.
In contrast our patients, who showed both a lower baseline MELD and lower rate of decompensated cirrhosis, did not experience serious AEs; no discontinuation of any of the study drugs was needed and only 11.7% required RBV dose reduction. Anemia was the most frequent AE but was low grade and easily managed without the need for red blood cell transfusion or erythropoietin support. Furthermore although the study population was smaller than the French study, there was no missing data, and all patients completed all scheduled visits.
A limitation of our study is the lack of randomization for treatment assignment regarding RBV, whereas treatment duration was the same in all patients. RBV was added based on physician's discretion, which can represent a possible bias. However RBV administration was discretionary by the treating clinician in all published studies of cirrhotic patients, due to variability of residual liver function across Child-Pugh classes and safety concerns regarding risk of decompensation [13–15]. The selection of patients who can benefit from RBV must take into account both pre-treatment SVR likelihood as well predicted tolerability. In the present study RBV was prescribed to patients with better residual liver function who were more likely to tolerate 24 weeks of treatment.
As the field of antiviral therapies is in continuous and accelerated development, shorter successful RBV-free regimens are now available with second generation DAAs, as reported in the ASTRAL-3 and POLARIS-3 trials [16,17]; a post hoc analysis of a second generation RBV-free DAA regimen based on SOF and velpatasvir (VEL) achieved high SVR rates, though extremely variable (86–98%) depending on patient characteristics especially among different stages of liver function [16,17]. In ASTRAL-4 decompensated cirrhosis patients infected with all genotypes were treated with 12 weeks of SOF+VEL with or without RBV, or with 24 weeks of SOF+VEL; addition of RBV increased SVR to a greater extent than 24 weeks’ duration, particularly in G3 infected patients, though in a small sample of patients (n=13) [18]. However, these regimens still need validation in real-life clinical practice studies, having been made available only recently.
Newer upcoming drug combinations such as glecaprevir/pibrentasvir or Mk3 have also shown promise in G3 infected patients, including cirrhotics [19,20]. Further clinical experience in larger cohorts of patients will be necessary to verify the safety profiles of these newer drug combinations.
In conclusion our data shows that the combination of SOF+DCV±RBV for 24 weeks in compensated G3 cirrhotic patients achieves excellent virological results, with a 98.1% SVR12 rate, and an excellent safety profile.
AbbreviationsHCV hepatitis C virus direct antiviral agent sustained virological response genotype hepatocellular carcinoma sofosbuvir ribavirin daclatasvir Agenzia Italiana del Farmaco (Italian Medicine Agency) ribonucleic acid polymerase chain reaction resistance-associated variants non-structural model for end-stage liver disease adverse event European Association for the Study of the Liver velpatasvir
None declared.
Financial supportNone declared.



