




Background. We previously identified miR-146b as being up-regulated during the development of hepatic fibrosis using deep sequencing technology and gene expression analysis. However, the roles and related mechanisms of miR-146b in hepatic stellate cells (HSCs), which are involved in fibrogenesis and fibrosis, have not been elucidated.
Results. We report that miR-146b expression was increased in TGF-β1-treated HSCs. TGF-β1 enhanced a-SMA and COL1A1 protein expression in HSCs and stimulated proliferation of these cells compared with cells transfected with inhibitor NC. Conversely, miR-146b knock-down decreased α-SMA and COL1A1 expression and inhibited HSC proliferation. In addition, we found that miR-146b specifically regulated the translation of Krüppel-like factor 4 (KLF4) by targeting its 3’ untranslated region. Forced expression of KLF4 inhibited TGF-β1-induced enhancement of α-SMA and COL1A1 expression in HSCs, as well as proliferation of these cells. Moreover, miR-146b expression was negatively associated with KLF4 expression but positively associated with expression of α-SMA and COL1A1 during hepatic fibrosis.
Conclusions. Our findings demonstrate the participation of miR-146b as a novel upstream effector of HSC activation via direct targeting of KLF4. Thus, targeted transfer of miR-146b into HSCs could be a useful strategy for the treatment of hepatic fibrosis.
Hepatic fibrosis is characterized by excessive production and deposition of extra-cellular matrix (ECM) components, resulting in destruction of normal liver architecture and impairment of hepatic function.1,2 Common outcomes of hepatic fibrosis include the development of liver cirrhosis and hepatocellular carcinoma.3–5 Following liver injury of any etiology, HSCs are transformed into myofibroblasts (activated HSCs), which express α-smooth muscle actin (α-SMA) and produce ECM.6,7 HSC activation has been described as an initial stage in the development of hepatic fibrosis.8,9 Several factors and related pathways involved in HSC activation have been identified, including transforming growth factor- β1 (TGF-β1),10,11 vascular endothelial growth factor (VEGF),12 the mitochondrial pathway of apoptosis,13 and mitogen-activated protein kinase (MAPK).14 However, the molecular mechanism that regulates HSC activation remains unclear.1
MicroRNAs (miRNAs) are a number of small non-coding RNA molecules, which usually negatively regulate gene expression. Regulation of gene expression by miRNAs occurs through direct induction of mRNA degradation or through translational repression by binding to the 3’ untranslated region (3’-UTR) of the mRNA of target genes.15 miRNAs are involved in various biological processes, such as activation, development, apoptosis, and proliferation.16 In our previous studies, we analyzed miRNA expression profiles in fibrotic and normal rat liver samples by using a deep sequencing approach.17 We reported that miR-19b decreased COL1A1 expression and HSC proliferation via targeting of GRB2.18 In addition, we observed up-regulated miR-146b expression in kinetics change in fibrotic liver tissue compared with that in the normal liver tissue.17 These findings suggest that altered miR-146b expression could be associated with fibrogenesis. However, more studies are required to clarify the mechanism of miR-146b in the development and progression of hepatic fibrosis.
In the present study, we show that TGF-β1 treatment of HSCs resulted in up-expression of miR-146b in parallel with enhanced HSC activation and proliferation. We validated the computational prediction of miR-146b binding to the 3’-UTR of KLF4. In vitro analyses revealed that down-regulation of KLF4 by TGF-β1 induced HSC activation and proliferation. In vivo experiments showed that miR-146b expression correlated with HSC activation. These findings demonstrated that miR-146b could be an important regulator of the HSC activation pathway that modulated hepatic fibrosis.
Material and MethodsHSC-T6 cell culture and activationThe rat cell line HSC-T6 was obtained from the Medical Research Center and Clinical Laboratory, Xiangya Hospital (Changsha, Hunan, China). Cells were cultured in Waymouth’s MB 752/1 medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), 100 units/ mL penicillin, and 100 mg/mL streptomycin (Invitrogen, Gibco Cell Culture, Portland, OR, USA). Cells were plated onto 6-well plates at 30% confiuence. Initially, cells were cultured with DMEM/F12 containing 10% FBS for 6 h. The medium was then replaced with DMEM/F12 without FBS to starve the cells for 12 h. Cells were then cultured with DMEM/F12 containing 5 ng/mL TGF-β1 (without FBS) for 48 h.19 All cells were cultured at 37 °C in a humidified incubator with 5% CO2.
Cell transfectionCell transfections were performed using a Lipofectamine 2000 Kit (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. Different miR-146b mimics, an miR-146b inhibitor (GeneCopoeia, Guangzhou, China), or a KLF4 ORF clone (Genechem, Shanghai, China) were introduced into HSC-T6 cells at a final concentration of 50 nM. Related negative controls for the miR-146b mimics and the inhibitor (GeneCopoeia, Guangzhou, China) were also used in the experiments. Transfected cells were harvested after 48 h incubation.
Gene expressionRNA isolation, reverse transcriptase (RT)-PCR, and quantitative RT-PCR of miR-146b were performed as previously described.17 Data were normalized to U6 spliceosomal RNA expression. Gene expression of KLF4, α-SMA, COL1A1, and GAPDH was analyzed using primers listed in table 1. Amplification of cDNA was performed using a THUNDERBIRD SYBR qPCR Mix (ToYoBo, Japan) and an Applied Biosystems 7500 Real-Time-PCR System. The mRNA level was normalized to that of GAPDH.
Sequences of real-time PCR primer used throughout.
| Primer | Sequence | Temperature | Direction | Length |
|---|---|---|---|---|
| TGFβ1-F TGFβ1-R | 5’-AGAAGTCACCCGCGTGCTA-3’ 5’-TGTGTGATGTCTTTGGIIIIGTCAT-3’ | 56 | Forward Reverse | 69 |
| KLF4-F KLF4-R | CTTTCCTGCCAGACCAGATG GGTTTCTCGCCTGTGTGAGT | 58 | Forward Reverse | 226 |
| α-SMA-F α-SMA -R | TGCCATGTATGTGGCTATTCA ACCAGTTGTACGTCCAGAAGC | 58 | Forward Reverse | 61 |
| COL1A1-F COL1A1-R | 5’-GCATCAGGGTTTCAGAGCA-3’ 5’-CGTTGGGTCATTTCCACAT-3’ | 60 | Forward Reverse | 164 |
| GAPDH-F GAPDH-R | 5’-CTTCCGTGTTCCTCCCC-3’ 5’-GCATCAAAGGTGGAAGAAT-3’ | 58 | Forward Reverse | 205 |
| miR-146b-F miR-146b-R | TGAGAACTGAATTCCATAGGCTGT GCTGTCAACGATACGCTACG | 65 | Forward Reverse | 85 |
| U6-F U6-R | CGCTTCGGCAGCACATATACTA CGCTTCACGAATTTGCGTGTCA | 61 | Forward Reverse | 89 |
Western blotting was performed to detect protein expression of KLF4, α-SMA and COL1A1 in HSC-T6 cells and rat liver tissue, as previously described.18 Equal amounts of protein were separated by 10% SDS-PAGE and electrotransferred to a PVDF membrane. Membranes were blocked with 5% milk in TBS for 2 h at room temperature and incubated overnight at 4 °C, using the following primary antibodies: KLF4 (1:1000; Abcam, Cambridge, MA, USA), α-SMA (1:50; Abcam, Cambridge, MA, USA), COL1A1(1:1000; Abcam, Cambridge, MA, USA), or GAPDH (1:50000; Abcam, Cambridge, MA, USA); respective HRP-conjugated secondary antibodies (1:5000) were next applied for 1 h at room temperature. Protein signals were densitometrically analyzed using Image J software (NIH, Bethesda, MD, USA), and the ratio of the protein of interest to GAPDH was determined.
Luciferase reporter assayHSC-T6 cells were grown to 70% confluence and cotransfected with a wild-type 3’-UTR reporter plasmid, miR-146b mimics/inhibitor, and pRL-TK plasmids. For negative control experiments, cells were transfected with a mutated KLF4 3’-UTR reporter plasmid, miR-146b mimics/inhibitor, and pRL-TK plasmids. Cells were harvested after 24 h, and the activities of both firefly and Renilla luciferases were measured using a LB 955 Luminometer system with a dual luciferase reporter system (Promega, Madison, WI, USA), according to the manufacturer’s instructions. The signal of firefly luciferase was normalized to that of Renilla luciferase.
Cell proliferation assayCell proliferation was analyzed using a Cell Counting Kit-8 (CCK-8, Beyotime, Shanghai, China), according to the manufacturers protocol. Briefly, cells were plated onto 96-well plates at a density of 5 × 103 cells/well and transfected with 5 ng/mL TGF-β1, 50 nM miR-146b inhibitor, or a KLF4 ORF plasmid, and negative controls. CCK-8 reagent (10 µL) was added at 1, 2, 3, and 4 d after transfection. After a 1-h incubation at 37 °C, the optical density (OD) was read at 450 nm by using a microplate luminometer reader.
Tissue samplesEight-week-old male Sprague-Dawley rats, weighing 120-150 g, were provided by the Department of Laboratory Animal Science, Xiangya Medical College, Central South University (Changsha, Hunan PR, China). Animals were maintained and experimental procedures performed in compliance with the Guide for the Care and Use of Laboratory Animals of the Chinese Academy of Sciences and approved by the Medicine Animal Care Committee of the First Affiliated Hospital of Nanchang University (Nanchang, Jiangxi PR, China). Twenty male rats were divided into normal control and fibrosis model groups (n = 10 per group). The fibrosis model group and the control group were maintained as previously described.18 At the end of week 12, rats were sacrificed under anesthesia using 10% chloral hydrate. Liver tissue was frozen after surgical removal and stored at −80°C.
StatisticsData were expressed as mean ± standard deviation from at least three independent experiments. Differences between two or more groups were evaluated using a Student’s t-test or a one-way analysis of variance (ANOVA). The association between miR-146b and KLF4, α-SMA and COL1A1 expression was determined using Spearman’s correlation analysis. P<0.05 was considered to be statistically significant. Statistical analysis was performed using SPSS 17.0 software (SPSS, Chicago, IL, USA).
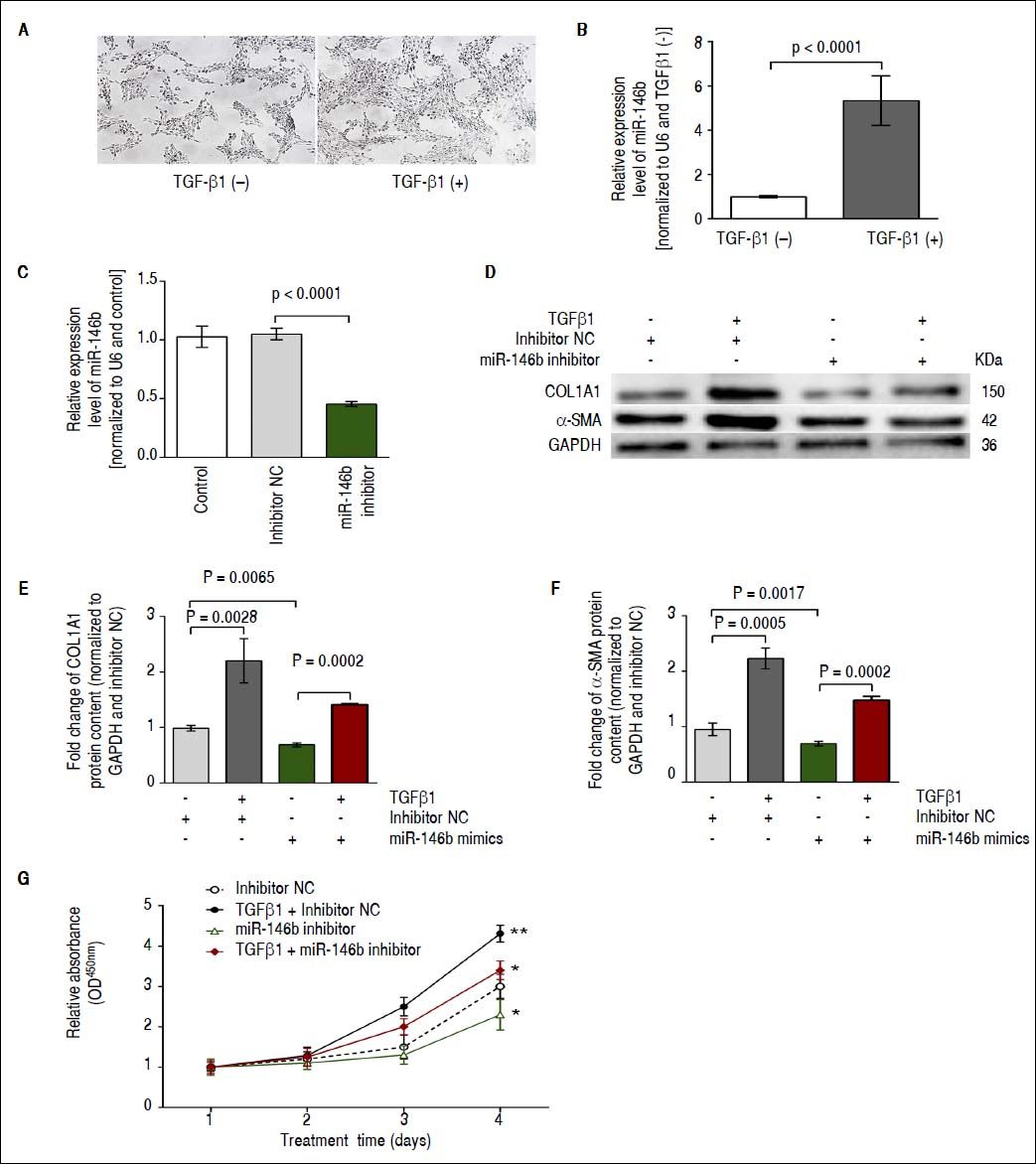
ResultsmiR-146b is up-regulated during TGF-β1-induced HSC activationWe previously identified miR-146b as being up-regulated during the development of hepatic fibrosis by using deep sequencing technology and qRT-PCR.17 TGF-β1 is a major profibrogenic protein induces HSC activation,20 and miR-146b has been shown to be responsive to this cytokine.21 Initially, cell morphology without and with TGFβ1 treatment showed that the density of HSC-T6 cells was increased in response to TGFβ1 stimulation, while no significant difference in the single morphology was observed (Figure 1A). We observed a significant increase in miR-146b expression in TGF-β1-stimulated HSCs, indicating that TGF-β 1 promoted miR-146b expression during HSC activation (Figure 1B). Moreover, we transfected HSCs with miR-146b inhibitor or inhibitor NC and observed that the miR-146b inhibitor down-regulated miR-146b expression (Figure 1C).

miR-146b is up-regulated during TGF-β1-induced HSC activation. A. Cell morphology without and with TGFβ1 treatment. B. Expression of miR-146b was induced by TGF-β1 treatment. C. miR-146b expression was evaluated in HSCs transfected with miR-146b inhibitor and HSCs transfected with an inhibitor NC using qRT-PCR. D. COL1A1 and α-SMA protein levels were evaluated by Western blot assays. E. COL1A1 protein content was enhanced by TGF-β1 treatment and inhibited by a miR-146b inhibitor; GAPDH was used as an internal control. F. Protein content of α-SMA was enhanced by TGF-β1 and inhibited by a miR-146b inhibitor; GAPDH was used as an internal control. G. HSC proliferation was increased by TGF-β1 and inhibited by a miR-146b inhibitor. Data are presented as the mean ± SD from three replicates in each group.
Next, we investigated the effects of TGF-β1 and miR-146b inhibition on HSC activation. Western blot results showed that in HSCs, TGF-β1 enhanced contents of α-SMA and COL1A1 compared with that of cells transfected with inhibitor NC; also, miR-146b knock-down reduced α-SMA and COL1A1 protein contents in TGF-β1-treated HSCs (Figure 1D-1F). A cell proliferation analysis showed that miR-146b knock-down in TGF-β1-treated cells inhibited cell growth compared with that of the control (Figure 1G). These data demonstrate that miR-146b promotes HSC activation and increases ECM protein production.
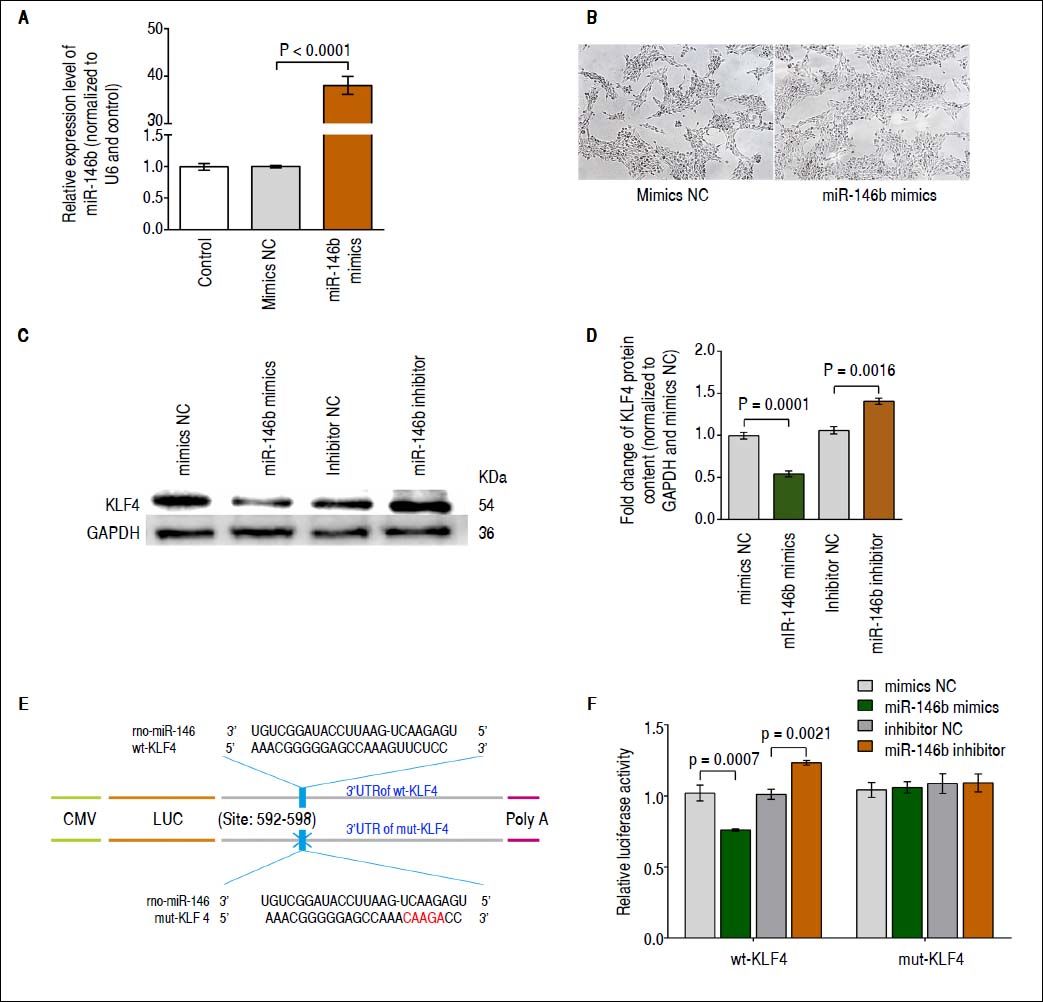
miR-146b inhibits KLF4 expression via targeting its 3’-UTRBased on several in silico analyses, including those performed using TargetScan 6.2,22 miRWalk,23 and miRanda,24 KLF4 was identified as a candidate gene potentially regulated by miR-146b. To determine whether miR-146b regulates KLF4 translation, we first generated HSCs in which miR-146b mimics or mimics NC were transiently over-expressed. Results obtained using qRT-PCR showed that at 48 h post-transfection, miR-146b expression was up-regulated compared with that of mimics NC (Figure 2A). In addition, miR-146b overexpression achieved by miR-146b mimics led to changes in the phenotype of cells. The density of HSC-T6 cells was increased in response to miR-146b overexpression, while no significant difference in the single morphology was observed (Figure 2B). Furthermore, KLF4 protein content was evaluated using Western blotting. Western blot analysis of these cells at 48 h post-transfection showed that miR-146b over-expression down-regulated endogenous KLF4 protein content compared with that of the control, whereas miR-146b inhibition up-regulated KLF4 protein content (Figures 2C and 2D). These findings indicate that KLF4 protein content is post-transcriptionally regulated by miR-146b.
 3’-UTR regions were obtained by co-transfection of a miR-146b mimics or miR-146b inhibitor, and pRL-TK plasmids; calculated values represent the ratio of firefly/Renilla luciferase activity normalized to that of the control. Data are presented as the mean ± SD from three replicates in each group.")
miR-146b inhibits KLF4 expression. A. miR-146b expression in HSCs transfected with miR-146b mimics and HSCs transfected with mimics NC was evaluated using qRT-PCR. B. Cell morphology without and with miR-146b overexpression. C. KLF4 protein levels were evaluated by Western blot assay. D. Protein content of KLF4 was decreased by miR-146b mimics compared with mimics NC group, and was increased by miR-146b inhibitor compared with inhibitor NC group, respectively; GAPDH was used as an internal control. E. The predicted miR-146b binding site within KLF4 3’-UTR and its mutated version by site mutagenesis were shown. F. Relative luciferase activity of wt-KLF4 and mutant (mut) 3’-UTR regions were obtained by co-transfection of a miR-146b mimics or miR-146b inhibitor, and pRL-TK plasmids; calculated values represent the ratio of firefly/Renilla luciferase activity normalized to that of the control. Data are presented as the mean ± SD from three replicates in each group.
To further validate the putative site of miR-146b initial binding within the 3’-UTR of KLF4 mRNA, we generated KLF4-3’-UTR luciferase reporter vectors containing the predicted wild type (wt) miR-146b binding site (wt-KLF4 3’-UTR) or a mutated version (mut-KLF4 3’-UTR) (Figure 2E). Next, the wt-KLF4 plasmid, miR-146b mimics/inhibitor, and the control were co-transfected into HSCs. The luciferase activity was reduced following transfection with miR-146b mimics and was induced by transfection with the miR-146b inhibitor but not by that of mutant KLF4 3’-UTR vector-transfected cells (Figure 2F). These results demonstrate that miR-146b directly inhibits KLF4 translation through a specific 3’-UTR mRNA binding sequence.
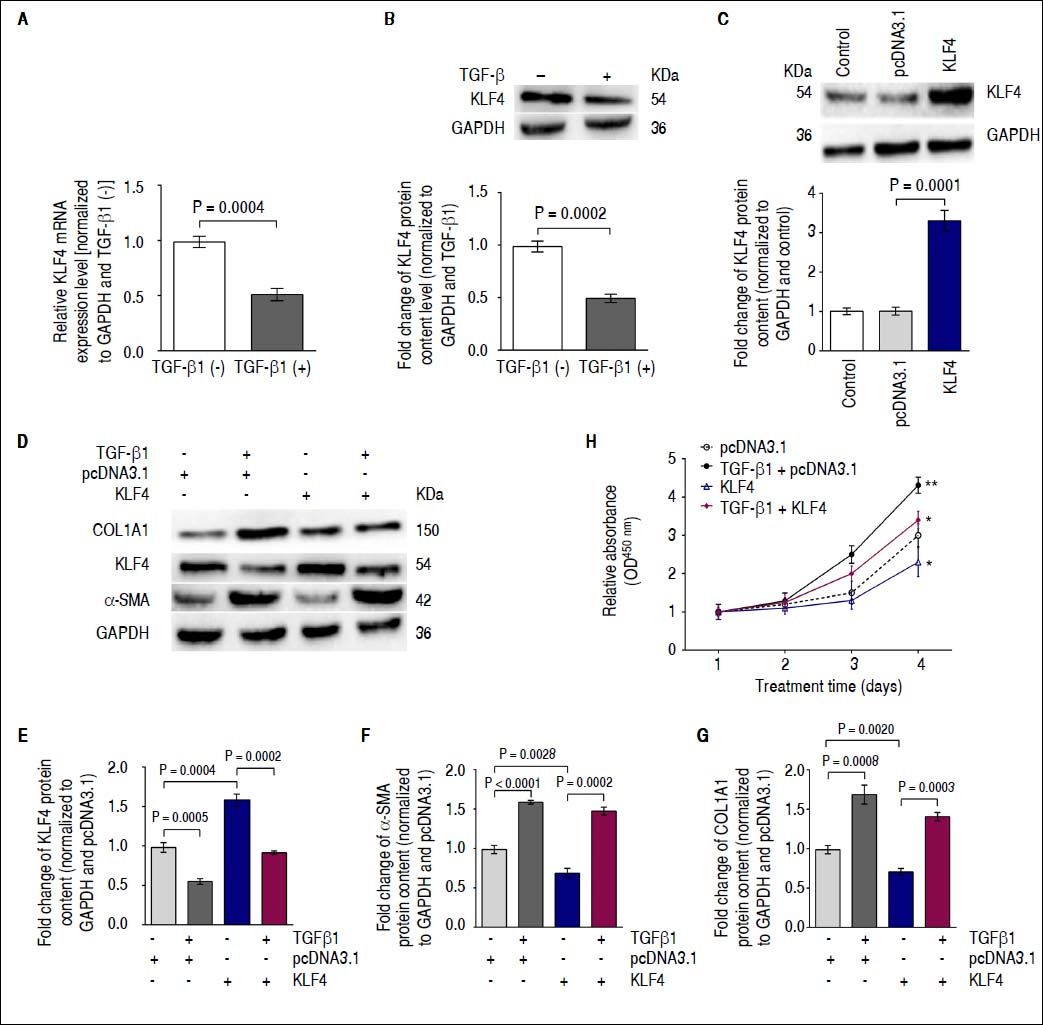
Down-regulation of KLF4 expression by TGF-β1 enhances α-SMA transcription in vitroIt was previously reported that KLF4 negatively regulates α-SMA expression25,26 and is inhibited by TGF-β1.27 Therefore, we investigated whether TGF-β1 regulates α-SMA transcription by down-regulating KLF4 in HSCs. Gene expression and immunoblotting results showed that TGF-β1 treatment on HSC resulted in decreased KLF4 mRNA expression and protein content compared with that of non-treated cells (Figures 3A and 3B). Furthermore, we transfected a KLF4 ORF clone into HSCs to up-regulate KLF4 protein content (Figure 3C). Next, we investigated α-SMA and COL1A1 protein levels using Western blotting analysis following co-transfection of TGF-β1 and the KLF4 ORF clone into HSCs. Western blotting results showed that forced expression of KLF4 inhibited the TGF-β1-induced increasing of α-SMA and COL1A1 protein content (Figures 3 D-G). Furthermore, we performed a proliferation assay by co-transfecting TGF-β1 and a KLF4 ORF clone into HSCs. Results showed that introduction of KLF4 into TGF-β1-treated cells, in which endogenous KLF4 was decreased, inhibited cell growth compared with that of TGF-β1-transfected cells (Figure 3H). Thus, the functional effects of TGF-β1 regulation of KLF4 are an enhancement of HSC activation and an increase in ECM protein synthesis.

Down-regulation of KLF4 expression by TGF-β1 leads to enhancement of α-SMA transcription in vitro. A. KLF4 mRNA expression was inhibited by TGF-β1 treatment. B. KLF4 protein levels were evaluated using a Western blot assay. KLF4 protein content was inhibited by TGF-β1 treatment. C. KLF4 protein content was up-regulated by transfecting a KLF4 ORF clone. D. KLF4, COL1A1 and α-SMA protein levels were evaluated using Western blot assays. E. Protein content of KLF4 was inhibited by TGF-β1 and enhanced by a KLF4 ORF clone; GAPDH was used as internal control. F. The protein content of α-SMA was enhanced by TGF-β1 and inhibited by a KLF4 ORF clone; GAPDH was used as an internal control. G. COL1A1 protein content was enhanced by TGF-β1 but inhibited by a KLF4 ORF clone; GAPDH was used as an internal control. H. The proliferation of HSC-T6 cells was increased by TGF-β1 treatment but inhibited by a KLF4 ORF clone. Data are presented as the mean ± SD from three replicates in each group.
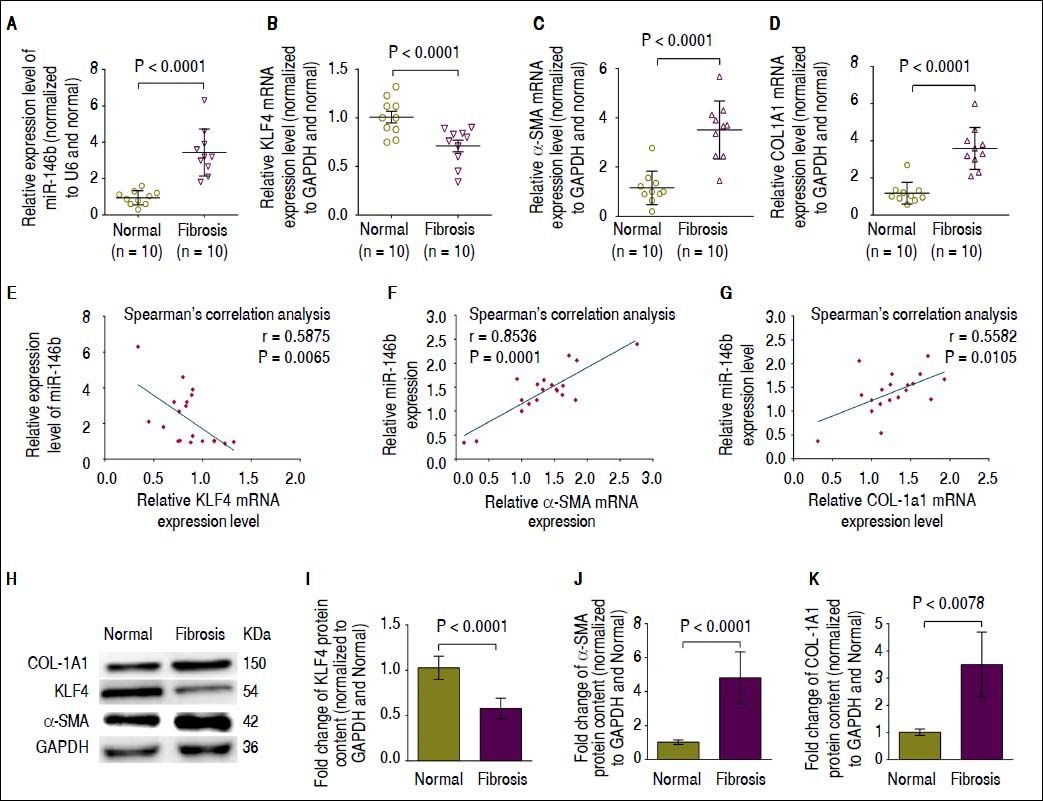
We next performed qRT-PCR and Western blot analysis to determine the mRNA expression and protein content of KLF4, α-SMA and COL1A1 in fibrotic liver tissue in which miR-146b expression was increased compared with that of normal liver tissue.17 Consistent with our previous report,17 miR-146b expression was much higher in fibrotic liver tissue compared with that in normal liver tissue (Figure 4A). Moreover, qRT-PCR and Western blot results showed that KLF4 mRNA expression and protein content was significantly decreased in fibrotic liver tissue compared with that in normal liver tissue (Figures 4B, 4H, 4I). Conversely, α-SMA and COL1A1 mRNA expression and protein content was significantly up-regulated in fibrotic liver tissue compared with that in normal liver tissue (Figures 4C, 4D, 4H, 4J, 4K). To determine whether miR-146b expression correlated with KLF4, α-SMA and COL1A1, we analyzed the relationship between miR-146b and KLF4, α-SMA and COL1A1 mRNA expression using Spearman’s correlation analysis. Spearman’s correlation analysis showed that miR-146b expression was inversely correlated with KLF4 mRNA (Figure 4E), but was positively correlated with α-SMA and COL1A1 mRNA expression (Figures 4F, 4G). These data suggested the possible involvement of miR-146b in the progression of liver fibrosis by negative regulation of KLF4 and positive regulation of α-SMA and COL1A1.

High miR-146b expression is correlated with down-regulation of KLF4 in vivo. A. Expression of miR-146b was up-regulated in fibrotic liver tissue compare with that in normal liver tissue. B. KLF4 mRNA expression was down-regulated in fibrotic liver tissue compared with that of normal liver tissue. C. The mRNA expression level of α-SMA was up-regulated in fibrotic liver tissue compared with that of normal liver tissue. D. COL1A1 mRNA expression was up-regulated in fibrotic liver tissue compared with that of normal liver tissue. E. Expression of miR-146b negatively correlated with KLF4 mRNA expression. F. Expression of miR- 146b positively correlated with α-SMA mRNA expression. G. Expression of miR-146b positively correlated with COL1A1 mRNA expression. H. KLF4, COL1A1 and α-SMA protein levels were evaluated by Western blot assays. I. KLF4 protein content was down-regulated in fibrotic liver tissue compared with that of normal liver tissue; GAPDH was used as an internal control. J. Protein content of α-SMA was up-regulated in fibrotic liver tissue compared with that of normal liver tissue; GAPDH was used as an internal control. K. COL1A1 protein content was up-regulated in fibrotic liver tissue compared with that of normal liver tissue; GAPDH was used as an internal control. Data are presented as the mean ± SEM from ten replicates in each group.
The trans-differentiation of HSCs into myofibroblasts is a pivotal process in the pathogenesis of hepatic fibrosis.28,29 miRNAs play an important role in liver disease by modulating HSC activation and proliferation.30,31 Microarray hybridization studies have identified 12 up-regulated miRNAs and nine down-regulated miRNAs that are differentially expressed during HSC activation.32 Furthermore, administration of miR-15b and miR-16 inhibited HSC proliferation and induced apoptosis by decreasing the expression of Bcl-2 but increasing that of caspases 3, 8, and 9.33 Additionally, over-expression of miR-181 greatly increased expression of α-SMA and further promoted HSC proliferation.19,34
We previously identified miR-146b as being up-regulated during the development of hepatic fibrosis using deep sequencing technology and gene expression analyses.17 miR-146b, located at chromosome 10, is characterized as a regulator of the inflammatory response.35,36 Recently, miR-146b was reported to be over-expressed in hepatic17,37 and cardiac fibrosis.38 Moreover, over-expression of miR-146b in fibroblasts contributed to the increased collagen content via its targeting of the tissue inhibitor of metalloproteinase (TIMP)-4.38 TGF-β1 is a major profibrogenic protein that induces HSC activation,20 and miR-146b has been shown to respond to this cytokine.21 Our results showed that TGF-β1 induced induction of α-SMA and COL1A1, as well as enhancement of HSC proliferation, occurred in parallel with miR-146b up-regulation (Figure 1). Thus, our findings provide evidence that miR-146b could be an important mediator of HSC activation.
As predicted by in silico analyses, molecular screens identified KLF4 as a candidate gene potentially regulated by miR-146b. We demonstrated a novel regulatory mechanism, in which miR-146b directly targeted KLF4, leading to increased α-SMA and COL1A1 production. In this regulatory network, miR-146b inhibited KLF4 expression by targeting its 3’-UTR (Figure 2). Moreover, forced expression of KLF4 inhibited the TGF-β1-induced enhancement of HSC proliferation as well as α-SMA and COL1A1 expression in these cells (Figure 3). These results demonstrate that miR-146b regulates HSC activation via the KLF4/α-SMA pathway.
KLF4 is a zinc-finger transcription factor implicated in pathological fibrogenesis. For example, mice with cardiomyocyte-specific deletion of KLF4 developed increased myocardial fibrosis.39 α-SMA is a key marker of HSC activation and the primary effector controlling the contractile potential of myofibroblasts.40–42 Expression of α-SMA is regulated by numerous factors, including TGF-β1.10,11 Previously, KLF4 was identified as a negative regulator of α-SMA expression by interfering with the binding of Smad2/3 to the α-SMA promoter.25,43,44 Recent studies have demonstrated that KLF4 over-expression results in decreased expression of α-SMA in renal fibrosis.25 It has been reported that miR-145 induced α-SMA expression through direct targeting of KLF4,26 which is an anti-proliferative regulator of vascular smooth muscle cells.45 Likewise, TGF-β1 induces activation of α-SMA expression in vascular smooth muscle cells, at least in part, by promoting KLF4 sumoylation and degradation.46 Moreover, enhanced KLF4 expression confers protection against lung fibrosis in miR-145-deficient mice.26
Our results showed that knockdown of miR-146b inhibited α-SMA and COL1A1 mRNA expression and protein synthesis in HSCs and decreased proliferation of these cells in association with KLF4 up-regulation. Thus, miR-146b could play a vital role in HSC activation by targeting KLF4. There are also recent data showing that KLF4 is a potential target gene of miR-146a,45 miR-145,26 and miR-10b.47 Ectopic miR-146b expression in HSCs decreased KLF4 levels. In addition, KLF4 was down-regulated by TGF-β1 in HSCs, resulting in enhancement of α-SMA and COL1A1 mRNA expression and protein synthesis, and promotion of HSC proliferation. These findings suggest that KLF4 was post-transcriptionally regulated by different miRNAs, including miR-146b.
It was also reported that miR-146b expression was significantly increased in fibrotic liver tissue, consistent with previous observations.17,37 We showed that in fibrotic liver tissue, KLF4 expression was significantly down-regulated, whereas expression of α-SMA and COL1A1 was up-regulated. KLF4 levels were negatively associated with miR-146b expression during hepatic fibrosis. In addition, miR-146b levels were positively associated with expression of α-SMA and COL1A1 during hepatic fibrosis (Figure 4). Thus, these results highlighted the importance of miR-146b-mediated HSC activation through targeting of KLF4 and presented miR-146b as a potential promoter of hepatic fibrosis.
In conclusion, we showed that miR-146b partly controls the KLF4/α-SMA pathway involved in HSC activation. Moreover, miR-146b could represent a novel therapeutic target for HSC activation and hepatic fibrosis. Further studies focusing on the in vivo functions of miR-146b, are required to clarify the significance of miR-146b-mediated regulation of hepatic fibrogenesis.
FundingThis study is supported by grants from the National Nature Science Foundation of China (No. 81660109) and the Province National Natural Science Foundation of JiangXi (20161BAB205232). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing InterestsThe authors declare that they have no competing interests.
AcknowledgementsWe are greatly indebted to the faculty and staff of the Central South University Xiangya Hospital, whose names were not included in the author list, but who contributed to this study.
Author ContributionsShanfei Ge and Tianxing Xiang performed the experiments and wrote the paper. Jinni He, Jinwen and He Xiaowei Wang coordinated the study. Fei Liu and Tianxing Xiang collected the samples and assisted with the experiments. Lunli Zhang and Jianping Xie conceived the study, designed the experiments, and revised the paper. All the authors have read and approved the final manuscript.



