



Alcoholic hepatitis (AH) is characterized by high morbidity and mortality. MicroRNA-494-3p is possibly involved in the regulation of cancers, but its role in AH has been rarely studied.
Materials and methodsAH mice model and primarily cultured mice hepatic stellate cells (HSCs) model were constructed. Levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were analyzed by ELISA. Expressions of miRNAs, HSC activation-related proteins and fibrosis-related protein were analyzed by qRT-PCR and Western blot. Cell counting kit, colony formation and flow cytometry assays were used to detect cell viability, proliferation and apoptosis, respectively. The relationship between TNF receptor-associated factor 3 (TRAF3) and miR-494-3p was predicted and verified by TargetScan and dual-luciferase assay, respectively. Results of the above experiments were verified by rescue experiments using TRAF3.
ResultsLiver damage and miRNA expression were observed in AH mice, and AST and ALT levels were increased in serum of AH mice. MiR-494-3p was reduced in AH liver tissues, and it decreased the levels of α-SMA and fibrosis-related proteins. HSCs were isolated, and activating HSCs or upregulating miR-494-3p had a regulatory effect on the levels of miR-494-3p, HSC activation-related proteins and fibrosis-related proteins as well as cell viability, proliferation and apoptosis. In addition, miR-494-3p targeted TRAF3 and inhibited TRAF3 expression, while overexpressed TRAF3 promoted TRAF3 expression and rescued the regulatory effect of miR-494-3p on the levels of related proteins as well as cell viability, proliferation and apoptosis.
ConclusionsThis study provided a novel mechanistic comprehension of the anti-fibrotic effect of miR-494-3p.
Alcoholic hepatitis (AH) is a serious liver disease with high morbidity and mortality [1], and can progress into cirrhosis and hepatocellular carcinoma [2]. According to statistics, 10-20% of AH patients developed into cirrhosis annually [2]. Acetaldehyde, reactive oxygen species, endotoxins and cytokines promote liver fibrosis, induce or inhibit liver regeneration, which has been well acknowledged to be the pathogenic mechanisms of AH [3–6]. Long-term heavy drinking can cause alcoholic liver disease, resulting in the gradual development of initial alcoholic fatty liver towards alcoholic liver fibrosis [7]. Hepatic stellate cells (HSCs) play a critical role in liver fibrosis [8]. They are in the quiescent state in normal liver, but are activated after liver damage [9]. Activated HSCs either resume the quiescent state or undergo apoptosis [10]. Therefore, controlling the activation of HSCs is a promising therapy to antagonize liver fibrosis.
MicroRNA (miRNA) is widely studied in molecular biology research. It can participate in epithelial-mesenchymal transition, HSC activation and myofibroblast apoptosis through transcriptional regulation of transforming growth factor beta (TGFβ) and other cytokines. MiRNA also plays a key role in the occurrence of fibrosis [11]. The role of miRNAs in chronic liver diseases that lead to liver fibrosis, such as nonalcoholic fatty liver disease, viral hepatitis and alcoholic liver disease, has received increasing attention [12–14]. As a class of endogenous targeting molecule, miRNAs can target HSCs to overcome the shortcomings of drugs, such as non-specificity and toxicity, and thus are conducive to developing new treating strategies for liver fibrosis.
MiR-494, which is encoded by a gene located on chromosome 14q32.31, is considered to have a tumor-suppressive function and can be detected in various cancer tissues, such as gastric cancer, cholangiocarcinoma and lung cancer tissues [15–17]. In hepatocellular carcinoma, overexpression of miR-494 enhanced sorafenib resistance via mTOR pathway activation [18]. Studies have found that miR-494-3p could be used as a potential biomarker for hepatocellular carcinoma [19], and high level of miR-494-3p in hepatocellular carcinoma was correlated with aggressive clinicopathological characteristics and was predictive of poor prognosis of HCC patients [20]. However, there is no study on the role of miR-494-3p in AH-induced liver fibrosis.
Tumor Necrosis Factor Receptor (TNFR) Related Factor 3 (TRAF3) is a new protein related to the intracellular cytoplasmic domain of CD40 and its viral mimics-Epstein-Barr virus latent membrane protein 1 (LMP1) [21,22]. TRAF3 is a one of the most versatile members in the TRAF family [23,24]. For example, TRAF3 limits osteoclast formation induced by TNF, which mediates inflammation and joint destruction in inflammatory diseases, including rheumatoid arthritis [25]; TRAF3 impact B cell metabolism and exerts powerful restraint upon B cell survival and activation [26]; hepatocyte TRAF3 promotes HFD-induced or genetic hepatic steatosis in a TAK1-dependent manner [27].
In the current study, we explored the potentially role of miR-494-3p in the development of AH with liver fibrosis. The findings provide a novel diagnostic and therapeutic target for treating liver fibrosis caused by AH.
Materials and methodsEthics statementFrom March 2017 to March 2019, 30 paired samples of AH liver tissues and normal liver tissues were collected from Yantai Affiliated Hospital of Binzhou Medical University. All patients had signed informed consent before the surgery. The study was approved by the Ethics Committee of Yantai Affiliated Hospital of Binzhou Medical University (No.YT2016070053), and the animal protocol was approved by the Institutional Review Board of Yantai Affiliated Hospital of Binzhou Medical University (2018031046-52).
Preparation of human tissue specimensIn this study, we obtained 30 paired samples of AH liver tissues and normal liver tissues from patients who were diagnosed with AH by pathological examination and healthy volunteers, respectively. All tissues were cut into about 1 cm3 [3] blocks with a sterile knife and washed twice with normal saline. Then, all tissues were immediately frozen in liquid nitrogen and transferred to a -80℃ refrigerator for preservation.
Establishment of a mice model of alcoholic hepatitisA total of 36 male C57BL/6 J mice, aged 7–8 weeks and weighing 20∼22 g, were obtained from Shilek Lab Animal (Shanghai, China). Animal license number: SYXK (Shanghai): 2019−0102. The animals were housed in normal pellets for 1 week at 22℃ under a 12 h/12 h light/dark cycle. In order to detect liver pathological changes and miRNA expression in AH mice, 12 of the mice were divided into Control group (n = 6, mice were fed with a control diet) and AH group (n = 6, mice were fed with a 4% alcohol Lieber-De Carli liquid diet (Hebei Hengshui Laobai Dry Wine Co., Ltd.) for 8 weeks). In order to further observe the effect of miR-494-3p mimic on AH mice, the remaining 24 mice were randomly divided into 4 groups, namely, Control group (n = 6), AH group (n = 6), AH + miR-494-3p mock (Mock) group (n = 6, mice were fed with a 4% alcohol Lieber-De Carli liquid diet for 8 weeks, and treated with tain vein injection of miR-494-3p mock since the 4th week), and AH + miR-494-3p mimic (Mimic) group (n = 6, mice were fed with a 4% alcohol Lieber-De Carli liquid diet for 8 weeks, and treated with tail vein injection of miR-494-3p mimic since the 4th week). Lieber-De Carli Alcohol liquid feeding consisted of 36% alcohol, 18% protein, 35% fat and 11% carbohydrate, which was modified from a previous report [28]. After 8 weeks, all mice were sacrificed by neck dislocation after being anesthetized with 0.2 mL of 1% pentobarbital sodium (P3761, Sigma-Aldrich, USA).
Preparation of serum samplesAll mice were fasted for 12 h after the last feeding, weighed, and anesthetized by intraperitoneal injection of 1% pentobarbital sodium. 3∼5 mL of blood was taken from the abdominal aorta of each mice and placed into a biochemical blood collection tube, and then centrifuged at 4 ℃, 1,006.2 ×g for 10 min (min). Serum was collected and stored at -80℃.
Enzyme linked immunosorbent (ELISA) assayAspartate aminotransferase (AST, EM0857) and alanine aminotransferase (ALT, EM0829) were purchased from FineTest (Wuhan, China). Following the instructions of the ELISA kit, the absorbance at 450 nm was determined by a microplate reader (MD SpectraMax M5, Molecular Devices, USA), and the expressions of ALT and AST in serum were analyzed according to the standard curve drawn by OD value.
Hematoxylin and eosin staining assayThe Haematoxylin-Eosin (HE) staining kit (XY0516 N) was obtained from Xinxu (Shanghai, China). The liver tissues were cut into 0.2∼0.3 cm thickness. After removing the surrounding adipose tissues, the liver tissues were fixed with 10% neutral formaldehyde solution (M004, G fan, Shanghai, Beijing), dehydrated with gradient alcohol, embedded in wax and dewaxed. Hepatic steatosis and inflammation in the liver sections were visualized by HE staining and observed under a dark field fluorescence microscope (DM2000, Olympus, Tokyo, Japan) under 100 × and 200 × magnifications.
ɑ-SMA immunohistochemical assayThe liver tissue sections were deparaffinized, hydrated and incubated in 3% hydrogen peroxide. The sections were then placed in 10 mM sodium citrate buffer (pH 6.0, Biomart, Beijing, China) and warmed in a microwave for 10 min for antigen retrieval. After 30 min of blocking in 0.1% Triton X-100 (DXT-11332481001, Roche, USA) at room temperature, the sections were incubated with primary antibody (Rabbit α-SMA antibody, 1:500, K10018, Biomart, Beijing, China) overnight at 4℃, followed by incubation with the secondary antibody Polymer-horseradish peroxidase anti-rabbit (Dako). After visualizing the proteins with 3,3’- diaminobenzidine, the sections were observed under a fluorescence microscope (DM2000, Olympus, Tokyo, Japan) under 100× and 200 × magnifications.
Isolation, culture and identification of primary hepatic stellate cellsC57BL/6 J mice were perfused in situ with EGTA and collagenase (Roche, Indianapolis, IN, USA) to obtain total liver cell suspension, and primary hepatocytes were obtained by Percoll density gradient centrifugation [19]. The cells were adjusted to a density of 3 × 106 cells/mL and seeded in a 25 cm2 plastic culture flask that contained 5 mL DMEM complete medium (10% FBS, Gibco, Life Technologies) and 1% penicillin/streptomycin (Gibco, Life Technologies) at 37℃. After activating hepatic stellate cells (HSCs), ɑ-SMA was substantially expressed and detected by immunofluorescence. HSCs were incubated with primary antibody (anti-α-SMA) at 4℃ overnight and subsequently stained with secondary antibody (Polymer-horseradish peroxidase anti-rabbit). Fluorescence positive expression changes of cells were measured under a fluorescence microscope (DM2000, Olympus, Tokyo, Japan) under 400 × magnification and images were collected. Next, quantitative real-time polymerase chain reaction (qRT-PCR) was used to identify the expressions of miR-494-3p and activation-related proteins in activated HSCs.
Cell groupingFirstly, to observe the effect of miR-494-3p on HSCs, the cells were divided into Blank group (untransfected), Mock group (transfected with mock) and Mimic group (transfected with mimic). Then, to further observe the effect of miR-494-3p and TNF receptor-associated factor 3 (TRAF3) on HSCs, the cells were divided into Mock + negative control (NC) group (transfected with mock + NC), Mock + TRAF3 group (transfected with mock + TRAF3), Mimic + NC group (transfected with mimic + NC) and Mimic + TRAF3 group (transfected with mimic + TRAF3).
Cell transfectionMiR-494-3p mimic (5′-UGAAACAUACACGGGAAACCUC-3′) and Mock (5′-ACAUCUGCGUAAGAUUCGAGUCUA-3′) were obtained from RiboBio (Guangzhou, China). The full length TRAF3 sequence synthesized by YouBia (Chongqing, China) was inserted into pcDNA3.1 vector (VT9221, YouBia, China) to obtained TRAF3 overexpression plasmid, and pcDNA3.1 empty vector was used as negative control (NC). HSCs were seeded into 6-well plates (5 × 104 cells/mL) and transfected with miR-494-3p mimic/Mock alone (50 nM) or in combination with TRAF3 overexpression plasmid/pcDNA3.1 empty vector (1 µg) using Lipofectamine 2000 reagent (11668027, Invitrogen, USA) according to the instructions. After 48 h (h) of transfection, the transfection rate was detected by qRT-PCR.
qRT-PCROne ml of Trizol lysate was added to lyse the HSCs and liver tissues for total RNA extraction. The supernatant was collected and added with 200 μL of chloroform for 5 min at room temperature, followed by centrifugation at 12,000 ×g for 15 min at 4℃. The supernatant was collected and transferred to a new centrifugation tube. After being added with 500 μL of isopropanol and kept at room temperature for 10 min, the supernatant was centrifuged at 12,000 ×g for 10 min at 4℃. The resulting supernatant was discarded and the precipitate was washed by 1 mL of 75% ethanol (anhydrous ethanol and DEPC treated water) once, and centrifuged at 7500 ×g at 4℃ for 5 min. After discarding the ethanol, the precipitate was properly dried and then dissolved in 25 μL of DEPC water, and subsequently the total RNA concentration was measured by Nandrop. The total RNA was extracted from Trizol for reverse transcription reaction. The reaction conditions were set as: at 42℃ for 10 min, at 95℃ for 15 min, and storage at 4℃. The qPCR experiment was conducted with SYBR Green PCR Master Mix (Roche, Basle, Switzerland) on a RT-PCR detection system (ABI 7500, Life Technology, USA) under the conditions as follows: pretreatment at 95℃ for 10 min, followed by 40 cycles of 94℃ for 15 s (s) and 60℃ for 1 min, finally at 60℃ for 1 min and preservation at 4℃. Referring to existing research, related miRNAs (miR-494-3p, miR-30e, miR-182, miR-378a-3p and miR-202-3p) were screened for analysis. miRNA was isolated from HSCs and liver tissues using a miRNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. cDNA was generated with the miScript II RT Kit (QIAGEN) and amplified by qPCR using the miScript SYBR Green PCR Kit (QIAGEN). The gene copy number of each sample was expressed by the Cq value, and the relative expression of the genes was determined by the 2ΔΔCq method [29]. Primers are listed in Table 1. miRNA expression levels were normalized to U6, and gene expression was normalized to GAPDH.
Primers for qRT-PCR.
| Genes | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| miR-494−3p | ATTGGAACGATACAGAGAAGATT | GGAACGCTTCACGAATTTG |
| COL-1 | ATGTCTGGTTTGGAGAGAGCA | GAGGAGCAGGGACTTCTTGAG |
| TRAF3 | CAAGTGCAGCGTTCAGACTC | GCAGCCATAGCGCTTAAAAC |
| TIMP-1 | GGCTGTGAGGAATGCACA | TGGAAGCCCTTTTCAGAGC |
| ɑ-SMA | TTCCTTCGTGACTACTGCTGAG | CAAT GAAAGATGGCTGGAAGAG |
| MMP-9 | CTTCAAGGACGGTTGGTACTG | GGAAGATGTCGTGTGAGTTCC |
| DDR2 | GTCTCAGGCTACGTTCAGATG | GGAATCAAGCCACTCACACAC |
| FN1 | TGGCAGTGGTCATTTCAGATGC | TTCCCATCGTCATAGCACGTTG |
| ITGB1 | CCGCGCGGAAAAGATGAAT | ATGTCATCTGGAGGGCAACC |
| Vimentin | AAGAACACCCGCACCAAC | GTAGTTGGCAAAGCGGTCA |
| GFAP | AGTGGCCACCAGTAACATGCAA | GCGATAGTCGTTAGCTTCGTGCTT |
| GAPDH | ACAGCAACA GGGTGGTGGAC | TTTGAGGGTGCAGCGAACTT |
| U6 | TGACCTGAAACATACACGGGA | TATCGTTGTACTCCACTCCTTGAC |
HSCs at 5 × 104 cells/mL were added into 96-well plates and incubated for 24 h. After 24, 48, 72 and 96 h of transfection treatment, CCK8 solution (Beyotime Institute of Biotechnology, Beijing, China) was added into the cells. After 2 h, the absorbance at 450 nm was measured using a microplate reader (MD SpectraMax M5, Molecular Devices, USA).
Cell clone formation experimentHSCs (5 × 104 cells/mL) were seeded into 6-well plates containing 37℃ pre-warmed medium and placed in a 37℃ incubator for culture. The medium was changed every two days and allowed to stand for 14 days. HSCs were fixed by 1:3 acetic acid/methanol for 30 min, and stained by a Giemsa stain (48900, Sigma-Aldrich, USA) for 20 min. The clone numbers of the cells were counted by naked eyes, and the colony formation rate was calculated using the equation: Clonal formation rate = (number of clones formed/number of cells seeded) × 100%.
Apoptosis analysisAn Annexin V-FITC/PI kit (CC2210, G-CLONE, Beijing, China) was used to evaluate the apoptosis of HSCs. HSC suspension at a final concentration of 1 × 106 cells/mL was prepared using 500 μL of 1× Annexin V Binding Solution, and then it was added to a 6-well plate. The cell suspension was added with 5 μL of Annexin V-FITC and 5 μL of propidium iodide and cultured in the dark for 15 min at room temperature. Then cell apoptosis was detected by Flow cytometry (version 10.0, FlowJo, FACS CaliburTM, BD, Franklin Lakes, NJ, USA). The necrotic cells were located in the upper left area (Annexin V−, PI+), and the late apoptotic cells were located in the upper right area (Annexin V+, PI+), while the living cells were located in the lower left area (Annexin V−, PI−), and the early apoptotic cells were located in the upper right area (Annexin V+, PI−).
Bioinformatics prediction and dual-luciferase reporter assayThe target gene of miR-494-3p was predicted using the internationally recognized prediction site TargetScan7.2 (http://www.targetscan.org/vert_72/). The mutant (Mut) and wild-type (WT) TRAF3 were amplified by PCR and cloned into pmirGLO reporter vector (E1330, Promega, USA) to generate TRAF3-WT and TRAF3-Mut report plasmids. Subsequently, cells were transfected with TRAF3-3'-UTR plasmid (TRAF3-WT and TRAF3-Mut) alone or in combination with miR-494-3p mimic using Lipofectamine 2000 reagent (11668019, Invitrogen, USA). After 48 h of transfection, the luciferase activity was measured using a luciferase reporter assay system (Promega Corporation) in Lmax II luminescence meter (Molecular Devices, LLC, Sunnyvale, CA, USA).
Western blot assayAccording to the literature [30], total proteins were extracted by RIPA lysate (PC901, Biomiga, USA). Total protein content was determined by a BCA Kit (93-K812-1000, Biovision, USA). The protein samples were separated by electrophoresis and then transferred to a membrane (PVDF, 2215, Millipore, CA, USA). The PVDF membrane was sealed with 5% skim milk at 37℃ for 1 h, and then separately incubated with Coll (1:2000 dilution, ab6308, Abcam, UK), matrix metalloproteinase 9 (MMP-9, 1:1000 dilution, ab38898, Abcam, UK), tissue inhibitor of metalloproteinase-1 (TIMP-1, 1:1000 dilution, ab61224, Abcam, UK), Vimentin (1:5000 dilution, ab92547, Abcam, UK) and GAPDH (1:10000 dilution, ab181602, Abcam, UK) at 4℃ overnight. Then goat anti-rabbit (1:5000 dilution, ab150077, Abcam, UK) or goat anti-mouse (1:5000 dilution, ab190475, Abcam, UK) was used to incubate the membrane at 37℃ for 1 h. Finally, immunoreactivity was detected with chemiluminescence reagent (PN3300, G-CLONE, Beijing, China), and color was developed in a gel imager (12003151, Bio-Rad, USA). GAPDH was used as a control.
Statistical analysisThe results were shown as the mean ± standard deviation (SD). Statistical significance was determined by analysis of variance (ANOVA) between groups followed by Bonferroni’s post hoc test using GraphPad Prism 7.0 (Graph-Pad Software Inc). Differences between two groups were compared by paired t test. P < 0.05 was considered as statistically significant.
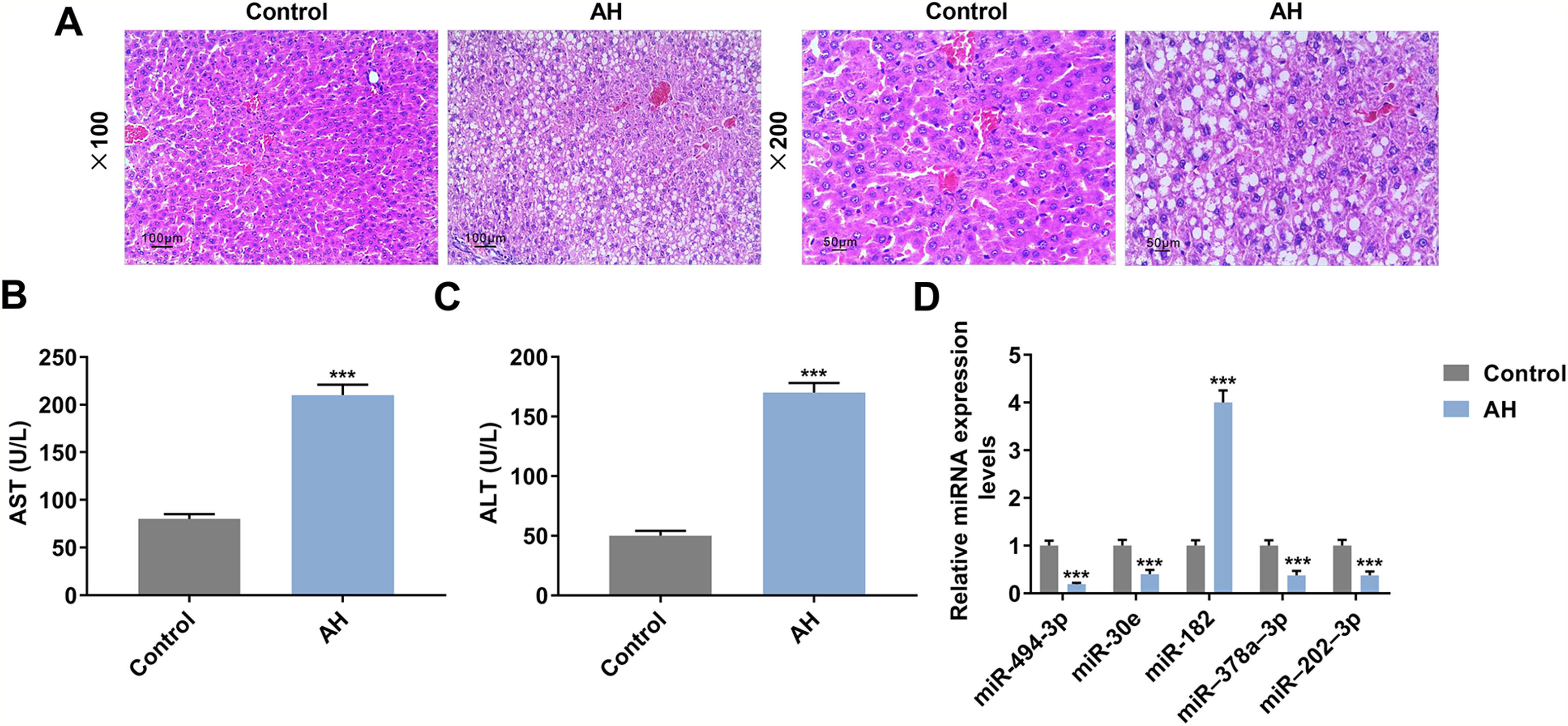
ResultsLiver damage and miRNA expression were observed in AH mice, and AST and ALT levels were increased in serum of AH miceAH mice model was successfully established, which was supported by pathological changes. Fig. 1A showed that hepatic lipid accumulation (predominantly macrovesicular) increased in AH mice. Moreover, chronic inflammatory cell infiltration was observed in AH mice compared with the control group (Fig. 1A). ELISA assay showed that AST and ALT levels were greatly enhanced in AH mice (P < 0.001, Fig. 1B and 1C). The results from qRT-PCR exhibited that the expression of miR-182 was greatly elevated in AH mice, while the expressions of miR-494-3p, miR-30e, miR-378a-3p and miR-202-3p were extremely reduced (P < 0.001, Fig. 1D), and therefore, miR-494-3p was selected for follow-up experiments.
. B-C. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels in AH mice were analyzed by enzyme linked immunosorbent (ELISA) assay. D. Expressions of miRNAs in AH mice were analyzed by quantitative real-time polymerase chain reaction (qRT-PCR). Expression levels were normalized to U6. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). ***P < 0.001 vs Control group.")
Liver damage and miRNA expression were observed in AH mice, and AST and ALT levels were increased in serum of AH mice.
A. Inflammatory cell infiltration in AH mice was observed by HE staining (magnification × 200 and × 100, scale bars = 100 μm). B-C. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels in AH mice were analyzed by enzyme linked immunosorbent (ELISA) assay. D. Expressions of miRNAs in AH mice were analyzed by quantitative real-time polymerase chain reaction (qRT-PCR). Expression levels were normalized to U6. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). ***P < 0.001 vs Control group.
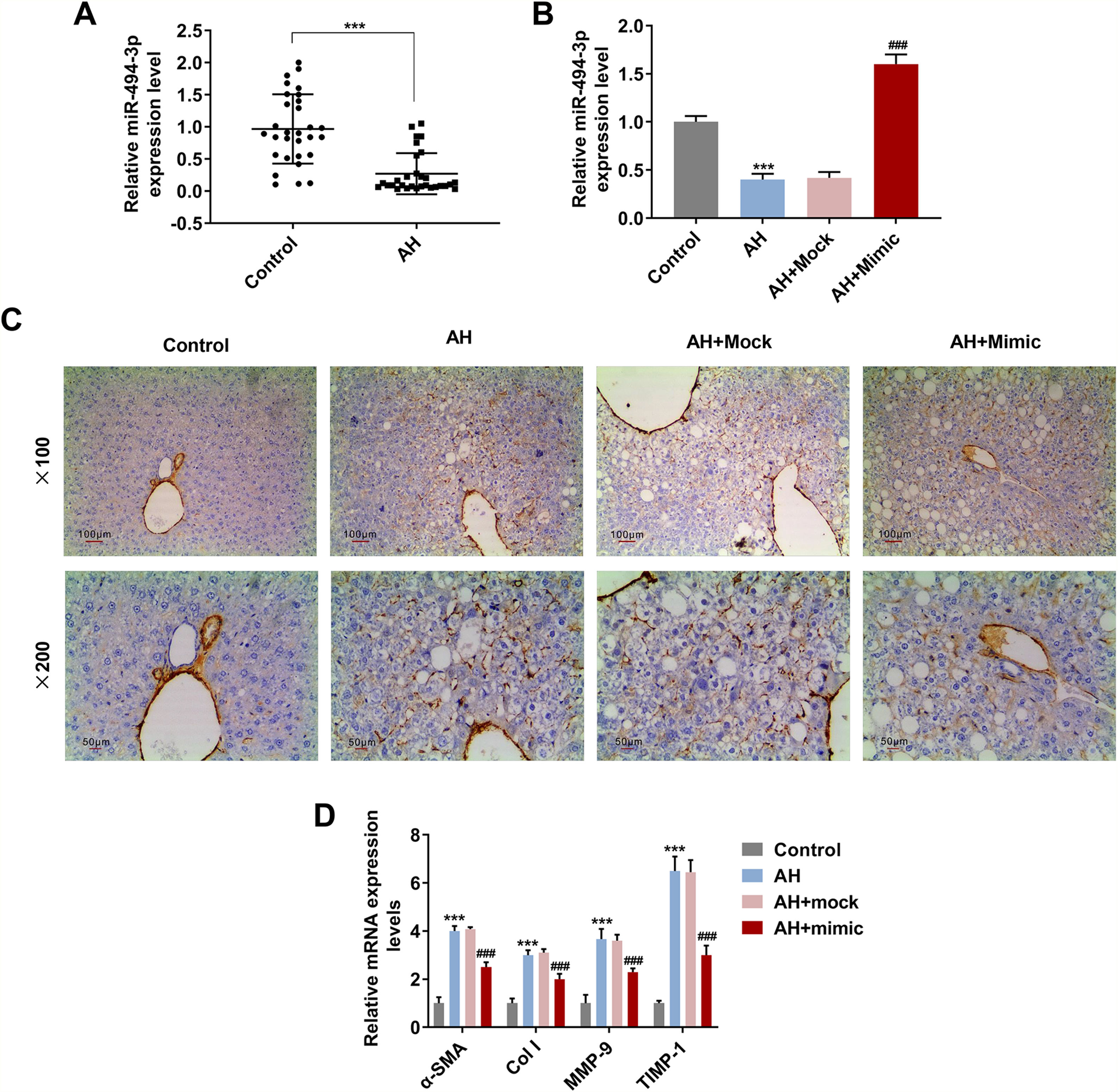
The mRNA level of miR-494-3p was visibly lower in human and mice AH liver tissues than in healthy volunteers’ liver tissues (P < 0.001, Fig. 2A and 2B), while miR-494-3p mimic obviously enhanced miR-494-3p level in AH mice (P < 0.001, Fig. 2B). Also, the expression of α-SMA was largely reduced in the AH + mimic group in comparison with the AH + Mock group (Fig. 2C). Moreover, the data from qRT-PCR showed that miR-494-3p mimic inhibited the mRNA levels of α-SMA, COL-1, MMP-9 and TIMP-1 in AH mice compared with the AH + Mock group (P < 0.001, Fig. 2D).
 patients and healthy volunteers was detected by quantitative real-time polymerase chain reaction (qRT-PCR). Expression levels were normalized to U6. B. Transfection efficiency of miR-494-3p mimic in AH mice was determined by qRT-PCR. Expression levels were normalized to U6. C. Immunohistochemical analysis of α-SMA expression in AH mice (magnification × 200 and × 100, scale bars = 100 μm). D. Eectopic expression of miR-494-3p suppressed the levels of fibrosis-related proteins in AH mice, as detected by qRT-PCR assay. Expression levels were normalized to U6. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). ***P < 0.001 vs Control group; ###P < 0.001 vs AH + miR-494-3p mock (Mock) group.")
MiR-494-3p was down-regulated in human and mice AH liver tissues, and it reduced collagen area and prevented fibrosis in AH mice.
A. The expression of miR-494-3p in liver tissues from alcoholic hepatitis (AH) patients and healthy volunteers was detected by quantitative real-time polymerase chain reaction (qRT-PCR). Expression levels were normalized to U6. B. Transfection efficiency of miR-494-3p mimic in AH mice was determined by qRT-PCR. Expression levels were normalized to U6. C. Immunohistochemical analysis of α-SMA expression in AH mice (magnification × 200 and × 100, scale bars = 100 μm). D. Eectopic expression of miR-494-3p suppressed the levels of fibrosis-related proteins in AH mice, as detected by qRT-PCR assay. Expression levels were normalized to U6. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). ***P < 0.001 vs Control group; ###P < 0.001 vs AH + miR-494-3p mock (Mock) group.
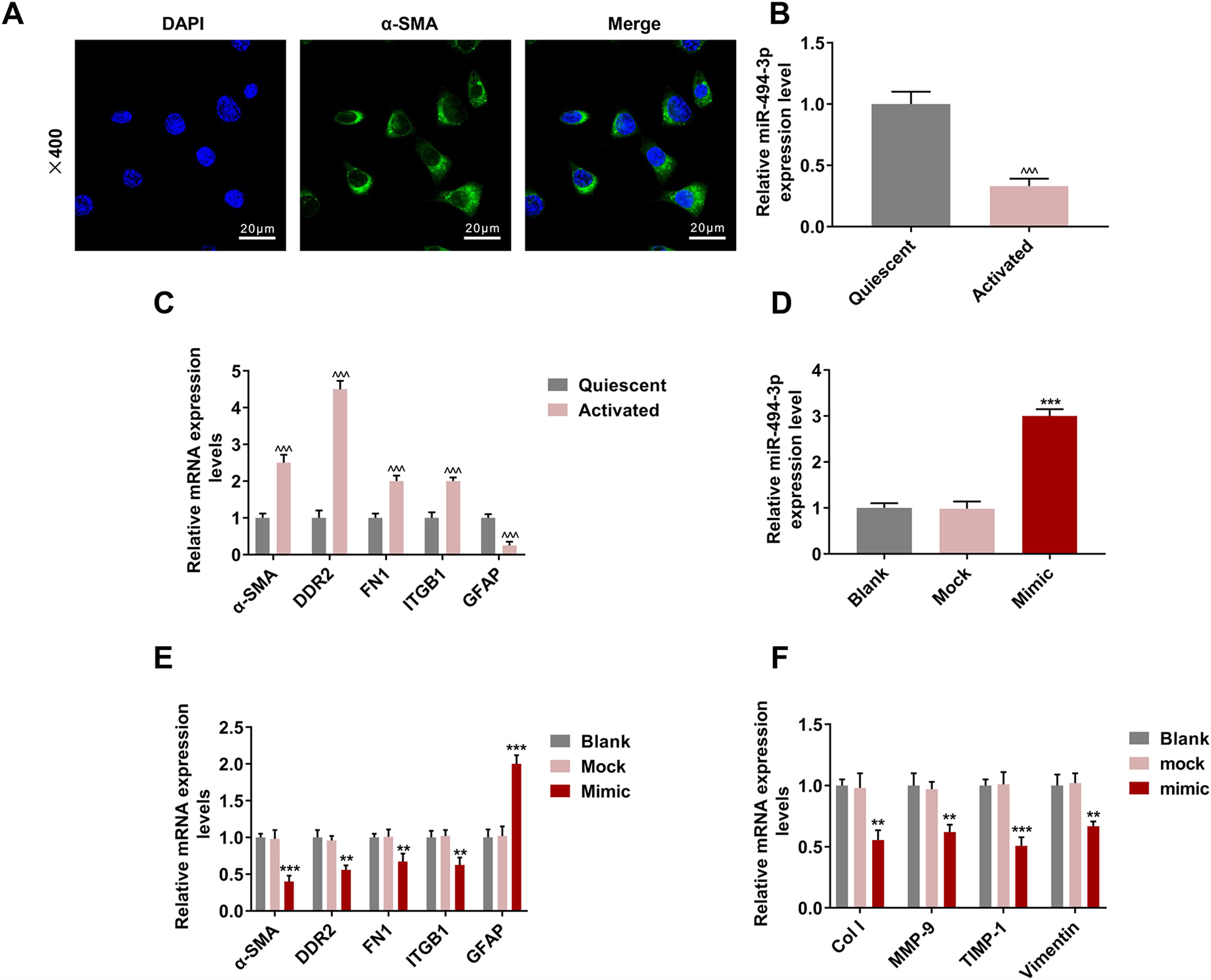
The positive expression of ɑ-SMA showed that HSCs were successfully isolated from the mice (Fig. 3A). We discovered that miR-494-3p was abundant in quiescent HSCs but decreased obviously in activated HSCs (P < 0.001, Fig. 3B). Furthermore, the levels of HSC activation-related proteins ɑ-SMA, DDR2, FN1 and ITGB1 were up-regulated, while GFAP expression was down-regulated in activated HSCs (P < 0.001, Fig. 3C). To determine the role of miR-494-3p in activated HSCs, miR-494-3p mimic was transfected into the cells to up-regulate the level of miR-494-3p, and the resulting changes were confirmed by qRT-PCR (P < 0.001, Fig. 3D). The mRNA levels of molecular markers ɑ-SMA, DDR2, FN1 and ITGB1 were down-regulated after transfection of miR-494-3p mimic, while GFAP expression was increased (P < 0.01, Fig. 3E). In addition, miR-494-3p mimic inhibited the mRNA levels of COL-1, MMP-9, TIMP-1 and Vimentin as compared with the Mock group (P < 0.01, Fig. 3F).
. B. MiR-494-3p level was down-regulated in activated HSCs, as detected by qRT-qPCR assay. C. Expression levels of activation-related proteins in HSCs were detected by qRT-qPCR assay. Expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). D. Transfection efficiency of miR-494-3p was up-regulated by miR-494-3p mimic, as detected by qRT-PCR assay. Expression levels were normalized to U6. E-F. The effect of miR-494-3p on the levels of HSC activation- and fibrosis-related protein was detected by qRT-PCR assay. Expression levels were normalized to GAPDH. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). **P < 0.01, ***P < 0.001 vs Mock group; ^^^P < 0.001 vs Quiescent group.")
HSCs were successfully isolated, and activating HSCs or upregulating miR-494-3p had a regulatory effect the levels of miR-494-3p and HSC activation-related proteins and fibrosis-related proteins.
A. Localization and expression of ɑ-SMA in cells were determined by immunofluorescence (magnification × 400, scale bars = 20 μm). B. MiR-494-3p level was down-regulated in activated HSCs, as detected by qRT-qPCR assay. C. Expression levels of activation-related proteins in HSCs were detected by qRT-qPCR assay. Expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). D. Transfection efficiency of miR-494-3p was up-regulated by miR-494-3p mimic, as detected by qRT-PCR assay. Expression levels were normalized to U6. E-F. The effect of miR-494-3p on the levels of HSC activation- and fibrosis-related protein was detected by qRT-PCR assay. Expression levels were normalized to GAPDH. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). **P < 0.01, ***P < 0.001 vs Mock group; ^^^P < 0.001 vs Quiescent group.
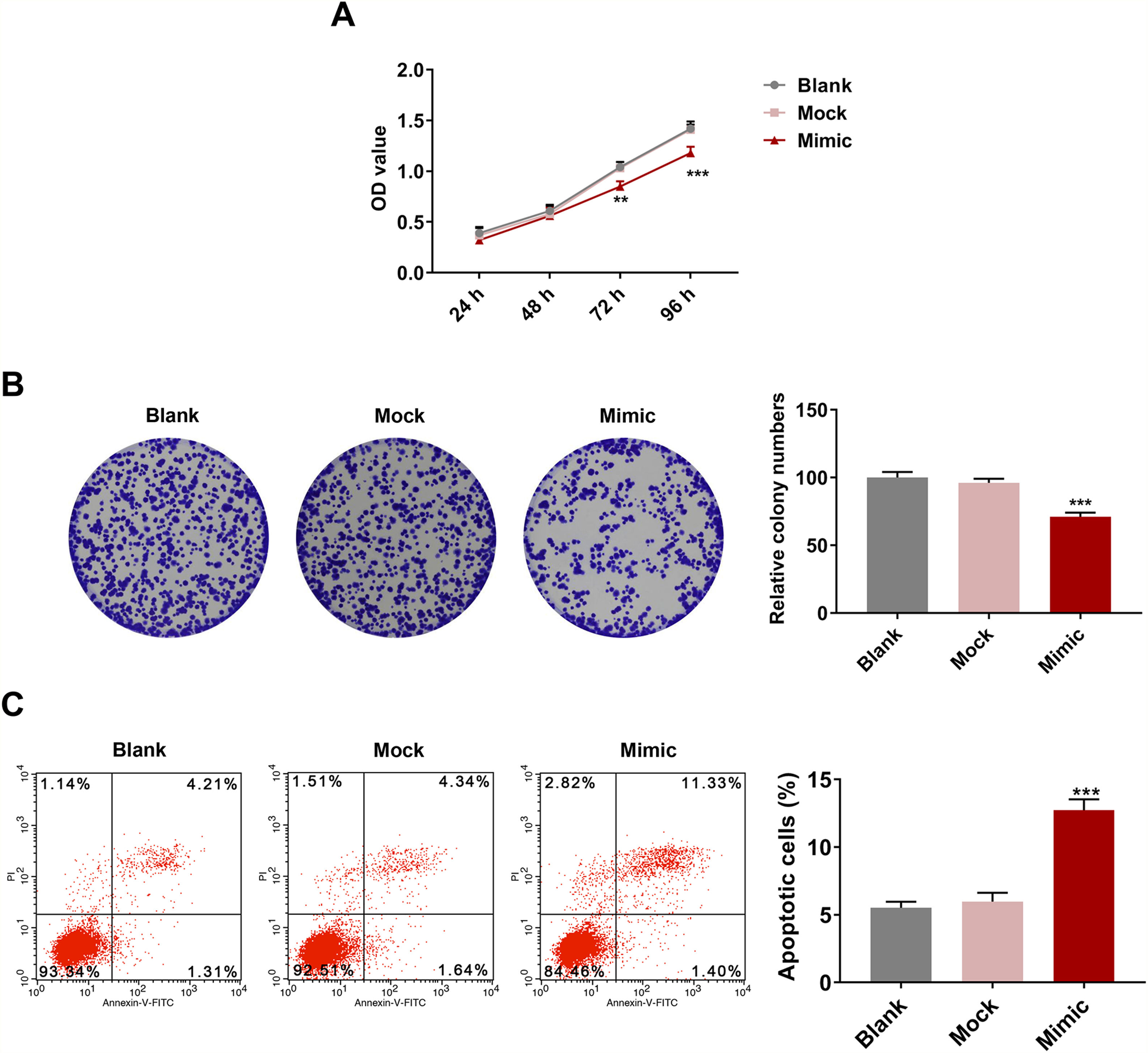
HSC viability was markedly inhibited after miR-494-3p mimic treatment for 72 h and 96 h (P < 0.01, Fig. 4A). Meanwhile, colony formation of HSCs was inhibited by miR-494-3p mimic (P < 0.001, Fig. 4B). We also found that HSC apoptosis was induced by miR-494-3p mimic (P < 0.001, Fig. 4C).
-8 assay showed that miR-494-3p inhibited HSC viability. B. Colony formation assay showed that miR-494-3p inhibited HSC proliferation. C. Flow cytometry assay showed that miR-494-3p induced HSC apoptosis. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). **P < 0.01, ***P < 0.001 vs Mock group.")
MiR-494-3p mimic inhibited viability and proliferation and induced apoptosis in HSCs.
A. Cell Counting Kit (CCK)-8 assay showed that miR-494-3p inhibited HSC viability. B. Colony formation assay showed that miR-494-3p inhibited HSC proliferation. C. Flow cytometry assay showed that miR-494-3p induced HSC apoptosis. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). **P < 0.01, ***P < 0.001 vs Mock group.
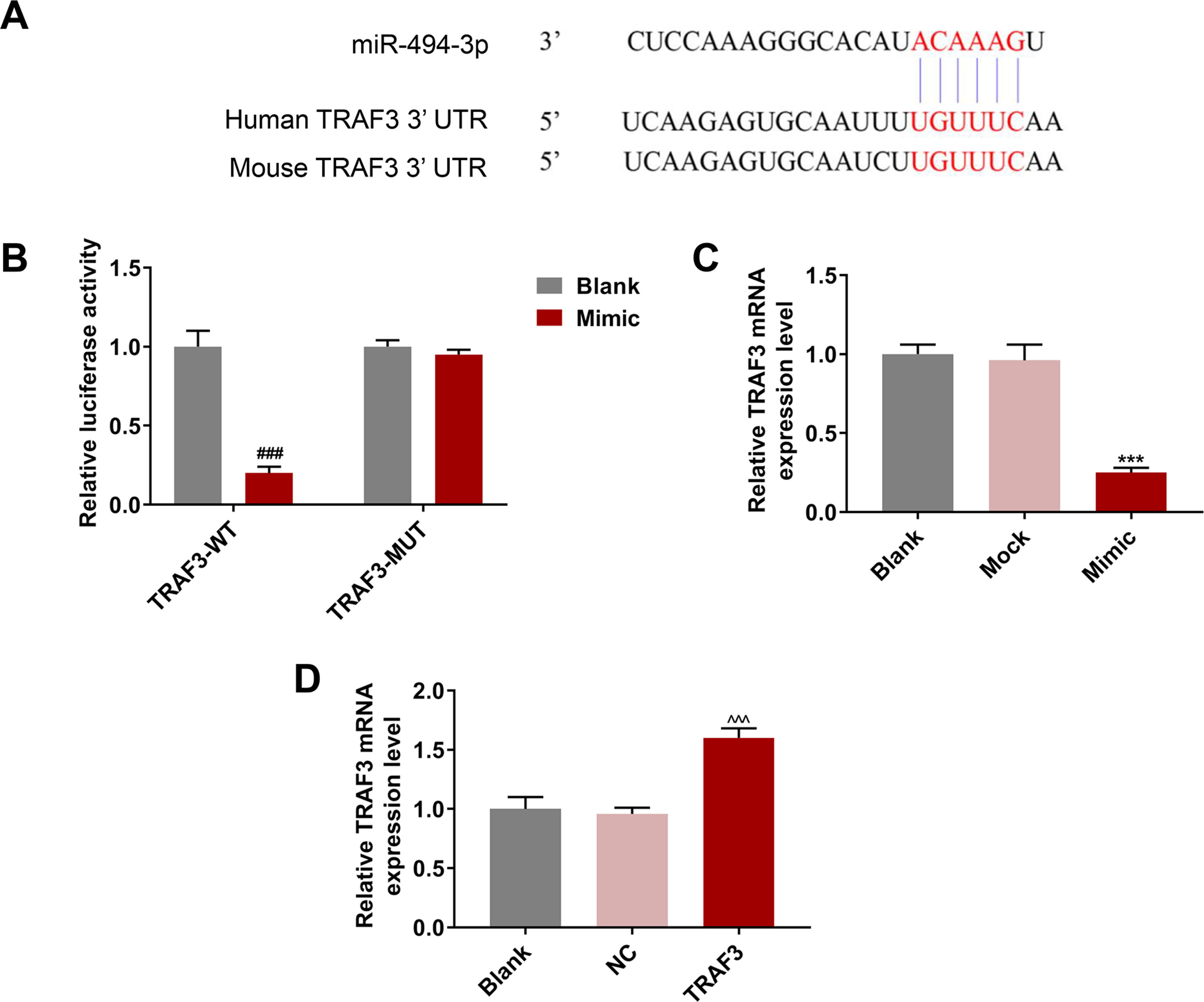
The target genes of miR-494-3p were predicted by TargetScan, and we found that the 3’-UTR of TRAF3 contained putative binding sites for miR-494-3p both in human and mice (Fig. 5A). Moreover, dual luciferase reporter assay showed that the luciferase activity of TRAF3-WT was inhibited by miR-494-3p mimic (P < 0.001, Fig. 5B). In addition, the effect of miR-494-3p on TRAF3 expression was detected, and the mRNA level of TRAF3 was decreased in the miR-494-3p mimic group compared with the mock group (P < 0.001, Fig. 5C). We found that TRAF3 level was increased in the TRAF3 group compared with the NC group (P < 0.001, Fig. 5D), which indicated that TRAF3 overexpression plasmid was successfully transfected.
 were predicted by TargetScan v7.2 (https://www.targetscan.org/). B. The direct interaction of miR-494-3p and TRAF3 was confirmed by dual-luciferase reporter assay. C. The effect of miR-494-3p on TRAF3 expression was determined by qRT-PCR assay. D. The transfection efficiency of TRAF3 was detected by qRT-PCR assay. Expression levels were normalized to GAPDH. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). ***P < 0.001 vs Mock group; ###P < 0.001 vs Blank group; ^^^P < 0.001 vs negative control (NC) group.")
MiR-494-3p targeted TRAF3 and inhibited TRAF3 expression, while overexpressed TRAF3 promoted TRAF3 expression.
A. The binding sites of miR-494-3p and TNF receptor-associated factor 3 (TRAF3) were predicted by TargetScan v7.2 (https://www.targetscan.org/). B. The direct interaction of miR-494-3p and TRAF3 was confirmed by dual-luciferase reporter assay. C. The effect of miR-494-3p on TRAF3 expression was determined by qRT-PCR assay. D. The transfection efficiency of TRAF3 was detected by qRT-PCR assay. Expression levels were normalized to GAPDH. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). ***P < 0.001 vs Mock group; ###P < 0.001 vs Blank group; ^^^P < 0.001 vs negative control (NC) group.
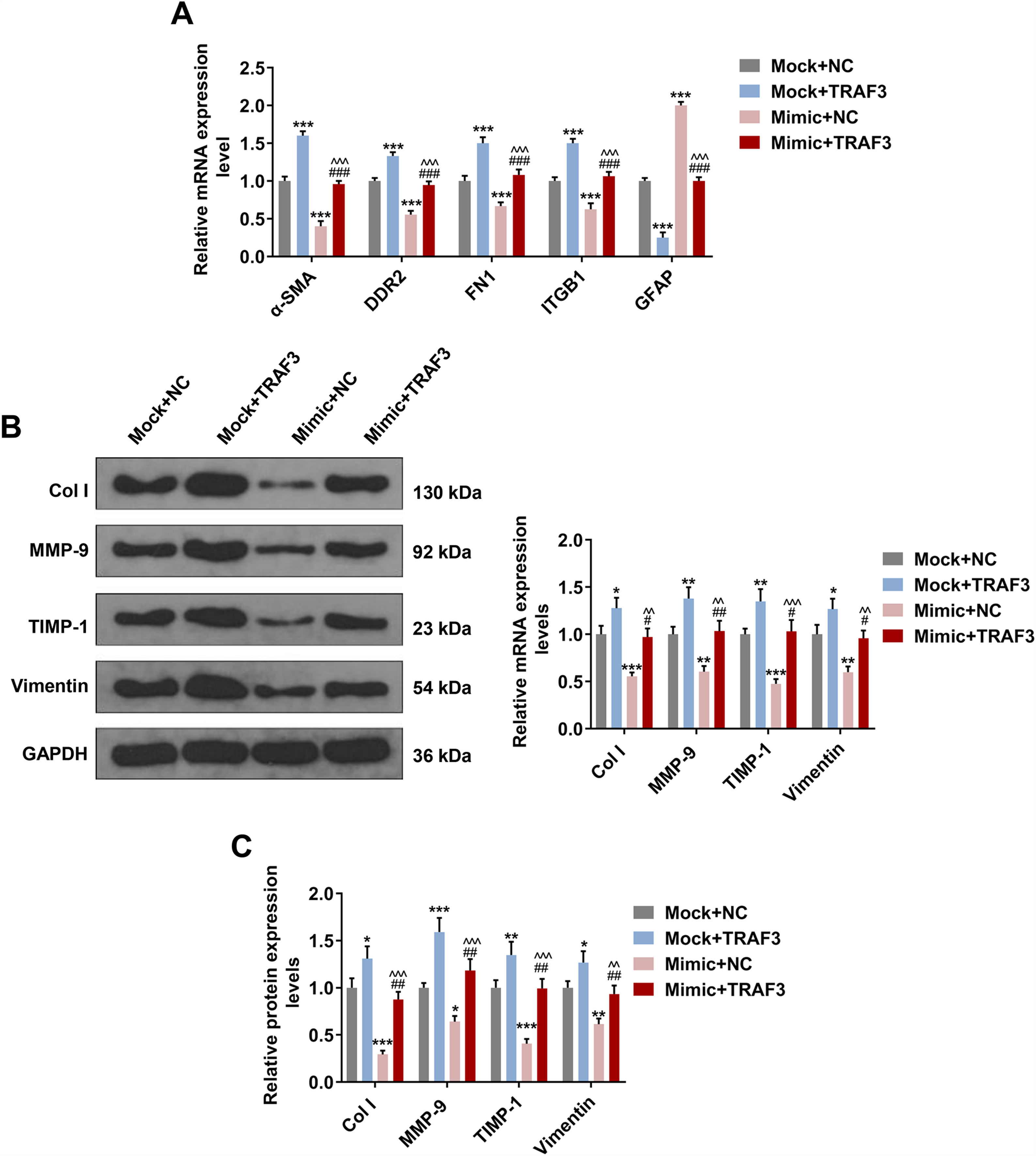
As depicted in Fig. 6A, the regulatory effect of miR-494-3p mimic on ɑ-SMA, DDR2, FN1, ITGB1 and GFAP expressions were reversed by overexpression of TRAF3 (P < 0.001, Fig. 6A). Furthermore, overexpressed TRAF3 partially offset the inhibitory effect of miR-494-3p mimic on the levels of fibrosis-related proteins, as evidenced by the enhanced mRNA and protein levels of COL-1, MMP-9, TIMP-1 and Vimentin (P < 0.05, Fig. 6B-C).
. *P < 0.05, **P < 0.01, ***P < 0.001 vs Mock + NC group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs Mock + TRAF3 group; ^^P < 0.01, ^^^P < 0.001 vs miR-494-3p mimic (Mimic) + NC group.")
Overexpressed TRAF3 rescued the regulatory effect of miR-494-3p mimic on the levels of HSC activation- and fibrosis-related protein.
A. The effects of miR-494-3p and TRAF3 on the levels of HSC activation-related protein were determined by qRT-PCR assay. Expression levels were normalized to GAPDH. B. The effects of miR-494-3p and TRAF3 on the levels of fibrosis-related proteins were determined by Western blot assay. Expression levels were normalized to GAPDH. C. The effects of miR-494-3p and TRAF3 on the levels of fibrosis-related proteins were determined by qRT-PCR assay. Expression levels were normalized to GAPDH. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001 vs Mock + NC group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs Mock + TRAF3 group; ^^P < 0.01, ^^^P < 0.001 vs miR-494-3p mimic (Mimic) + NC group.
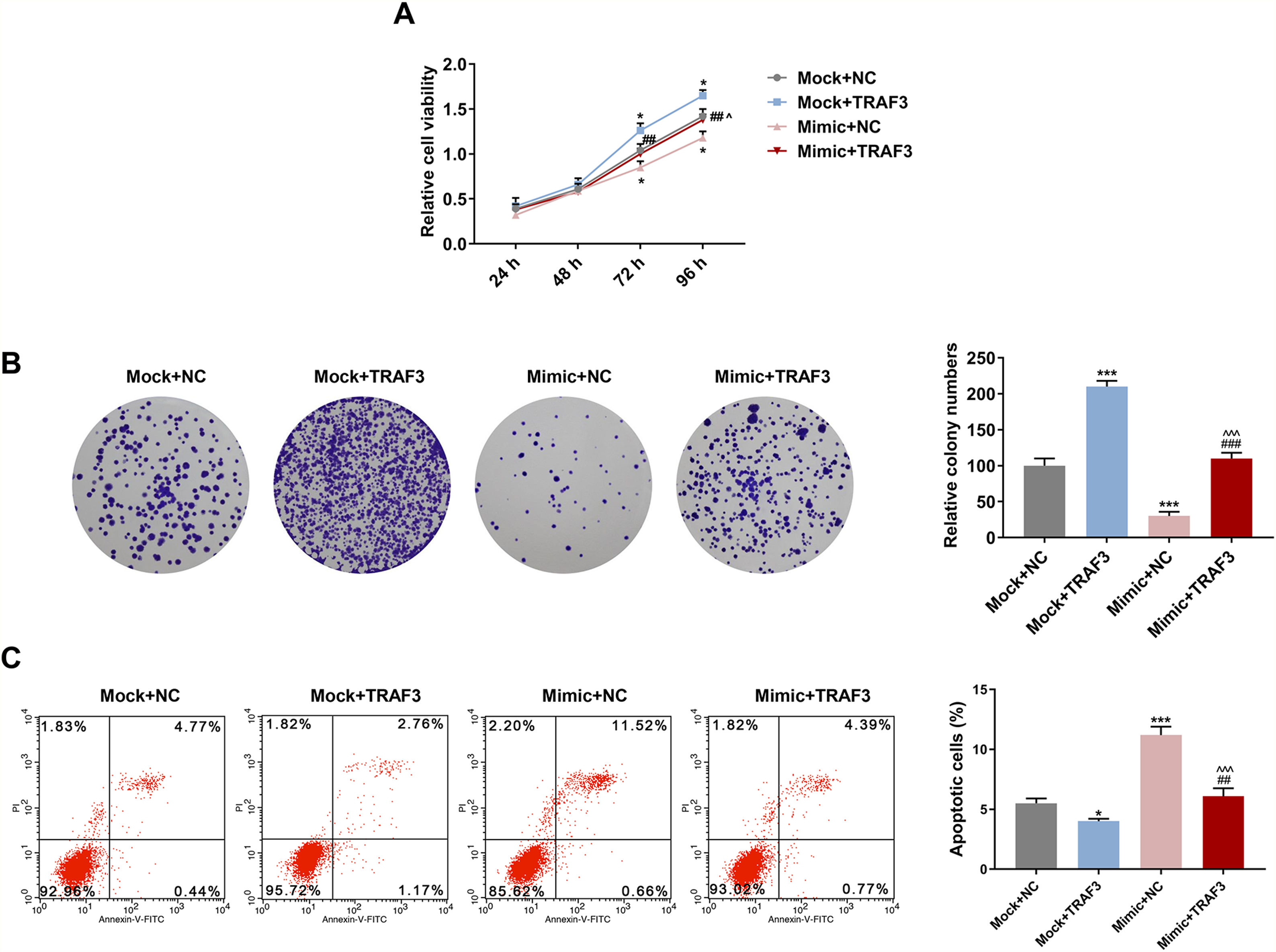
The CCK-8 data demonstrated that HSC viability was significantly enhanced after treatment with overexpressed TRAF3 for 72 h and 96 h (P < 0.05, Fig. 7A). Meanwhile, the inhibitory effect of miR-494-3p mimic on cell viability was reversed after treatment with overexpressed TRAF3 for 72 h and 96 h (P < 0.05, Fig. 7A). Moreover, introduction of TRAF3 notably increased colony numbers compared with transfection of miR-494-3p mimic alone (P < 0.001, Fig. 7B). In addition, Flow cytometry results revealed that overexpressed TRAF3 rescued the promoting effect of miR-494-3p mimic on the apoptosis of HSCs (P < 0.001, Fig. 7C).
. *P < 0.05, ***P < 0.001 vs Mock + NC group; ##P < 0.01, ###P < 0.001 vs Mock + TRAF3 group; ^P < 0.05, ^^^P < 0.001 vs Mimic + NC group.")
Overexpressed TRAF3 partially reversed the regulatory effect of miR-494-3p mimic on cell viability, proliferation and apoptosis.
A. The effects of miR-494-3p and TRAF3 on cell viability were determined by CCK-8 assay. B. The effects of miR-494-3p and TRAF3 on proliferation were determined by colony formation assay. C. The effects of miR-494-3p and TRAF3 on apoptosis were detected by flow cytometry assay. All experiments have been performed in triplicate and data were expressed as mean ± standard deviation (SD). *P < 0.05, ***P < 0.001 vs Mock + NC group; ##P < 0.01, ###P < 0.001 vs Mock + TRAF3 group; ^P < 0.05, ^^^P < 0.001 vs Mimic + NC group.
In the current study, our findings suggested that miR-494-3p mRNA level was down-regulated in AH liver tissues, and miR-494-3p mimic significantly reduced the expression of α-SMA and prevented liver fibrosis was prevented. In addition, we found that the expression and biological functions of miR-494-3p were regulated by TRAF3. Studies reported that miR-494-3p has low expression in various diseases, and moreover, it was reported as a novel noninvasive biomarker for hepatocellular carcinoma [20,31]. However, it is unclear whether miR-494-3p expression is associated with AH. In the present study, miR-494-3p level was down-regulated in liver tissues from AH patients and activated HSCs, thus suggesting that miR-494-3p was involved in AH development.
Many studies have shown that miRNAs play key roles in alcoholic hepatitis and fibrosis. For example, miR-378 limits liver fibrosis and HSC activation [32]. MiR-29b attenuates hepatic stellate cell activation and induces apoptosis against liver fibrosis [33], and miR-26b-5p suppresses angiogenesis and liver fibrogenesis in mice [34]. Additionally, miR-126 inhibits the activation and migration of HSCs through targeting CRK [35]. To better understand the biological function of miR-494-3p in AH, we transfected overexpressed miR-494-3p into AH mice, and the results from in vivo functional experiments showed that miR-494-3p mimic alleviated collagen deposition and fibrosis, suggesting that miR-494-3p may inhibit AH development in mice. Liver fibrosis is characterized by excessive deposition of extracellular matrix (ECM) components, particularly type I collagen [36]. After liver injury, HSCs change from a resting phenotype to an activated phenotype, migrate to the injured area, and produce ECM [37]. MMP-9 is one of the most relevant MMPs that degrades normal liver matrix, and it could promote the development of liver fibrosis [38]. TIMP1, which has been demonstrated to reduce MMP activity, plays an important role in the progress of liver fibrosis and is an important target for the treatment of liver fibrosis [39]. The α-SMA, COL-1, MMP-9 and TIMP-1 genes are mainly produced by HSCs during fibrogenesis [33]. In this study, miR-494-3p overexpression inhibited the expressions of these fibrosis-related proteins in AH mice, indicating that miR-494-3p could inhibit liver fibrosis.
When liver fibrosis is prevented, activated HSCs either keep a quiescent state or undergo apoptosis, and the latter leads to a decreased number of activated HSCs [10,40]. Down-regulation of miR-140-3p suppresses fibrogenesis and cell proliferation in HSCs [41]. MiR-193a/b-3p limits proliferation, relieves hepatic fibrosis and activates HSCs [42]. MiR-29b induced apoptosis of HSCs by regulating PARP and casepase-9 [33]. We found that miR-494-3p mimic inhibited the activation, proliferation and fibrosis of HSCs, indicating that up-regulation of miR-494-3p could inhibit liver fibrosis by inhibiting the proliferation of HSCs.
Our data confirmed that miR-494-3p targeted TRAF3. Studies have shown that TRAF3 regulates the homeostasis of various cell types through different mechanisms [43,44]. MiR-107 modulates the apoptosis and autophagy of osteoarthritis chondrocytes by regulating TRAF3 [45]. MiR-155-5p is negatively correlated with acute pancreatitis and inversely adjusts the development of pancreatic acinar cells by modulating TRAF3 [46]. This study found that TRAF3 is a target gene of miR-494-3p, and the protective effect of overexpressed miR-494-3p on HSCs could be offset by TRAF3. These findings indicated that miR-494-3p may inhibit HSC proliferation and fibrosis via regulating TRAF3.
To conclude, we proved that miR-494-3p suppressed HSC proliferation and fibrosis in AH by blocking TRAF3. Thus, our findings provide new treatment strategies for AH.
FundingThis work was supported by the Research on Non-invasive Diagnosis Method of OBI based on PBMC.
Ethics statementFrom March 2017 to March 2019, 30 AH liver tissues and normal liver tissues were collected from Yantai Affiliated Hospital of Binzhou Medical University. All patients had signed informed consent before the surgery. The study was approved by the Yantai Affiliated Hospital of Binzhou Medical University Ethics Committee (No.YT2016070053), and the protocol on animal was approved by the Institutional Review Board of the Yantai Affiliated Hospital of Binzhou Medical University (2018031046-52).
Declarations of interestNone.
Not applicable.



