



Currently, chronic liver diseases have conditioned morbidity and mortality, many of these with a metabolic, toxicologic, immunologic, viral, or other etiology. Thus, a transcription factor that has been of huge importance for biomedical research is NRF-2. The latter is considered a principal component of the antioxidant mechanism, and it has been acknowledged that it impairs the function of NRF-2 in many liver diseases and that it forms an essential part of the pathologic changes that occur in the liver to contain inflammation and damage. Within the investigations and experiments carried out, there are isolated drugs, many of them related to plants and natural extracts that possess antioxidant properties through the NRF-2 signaling pathway, or even involving the stimulation of the transcription target proteins of NRF-2. Notwithstanding all of these experimental findings, to date there is not sufficient clinical evidence to justify the use of NRF-2 in medical practice.
Non-alcoholic fatty liver disease is the most common chronic hepatic disease at the worldwide level [1,2], and is defined as increased intrahepatic fat accumulation, in the absence of risk factors for liver disease, such as alcohol abuse, steatogenic medications, and other causes of chronic liver disease [2]. According to results published by Younossi et al. in 2016, it has a global prevalence of 25.24% (95% CI: 22.10–28.65), where South Africa and the U.S. Midwest are the regions with the highest prevalence [3]. The youngest population is severely affected, with insulin resistance as a common proinflammatory condition in obesity [5]. The body mass index (BMI) had a significant correlation with the increase TNF-α and C-reactive protein [4].
Nuclear factor (erythroid-derived 2)-like 2 (NRF-2) is a transcription factor first identified by Moi et al. in 1994 [6]. To date, the modulation of proteins and enzymes against oxidative stress and inflammation is known as its main action, by means of activating the transcription of antioxidant response elements (ARE). In its inactive state, it interacts by its Neh2 (NRF2-ECH homology-2) domain with Kelch-like ECH associating protein-1 (KEAP1); as long as it is not exposed to reactive oxygen species (ROS), it is degraded by the ubiquitin-proteasome pathway [7]. When NRF-2 is exposed to inflammation or oxidative stress (OS), it phosphorylates and translocates into the nucleus, giving rise to the transcription of proteins and antioxidant enzymes [7]. The NRF-2 pathway aids the antioxidant mechanism with necessary compounds to protect against the OS, which will not be found in excessive situations and which can be maintained under control [8,9].
Continuous liver damage due to the overproduction of ROS and inflammation by diverse endogenous and exogenous injuries, failure at the respiratory chain in mitochondria [10,11], drop-in antioxidant elements such as superoxide dismutase [12], catalase activity [13], or the elevation of lipid peroxidation [14], among other described mechanisms, working together, are involved in injuries, apoptosis, inflammation, and fibrosis [10]. As mentioned by Wei Tang, 2014, oxidative stress and lipid peroxidation are the main components in the development of cirrhosis and hepatocarcinoma [8,9]. In a study by Gupte et al. in 2013, the authors reported the decrease of the activity by NRF-2 in liver tissue accompanied by the criteria of nonalcoholic steatohepatitis (NASH) [15].
The purpose of the following review is to present the most recent advances in the physiological, immunological, and therapeutic properties in patients with nonalcoholic fatty liver disease and NRF-2.
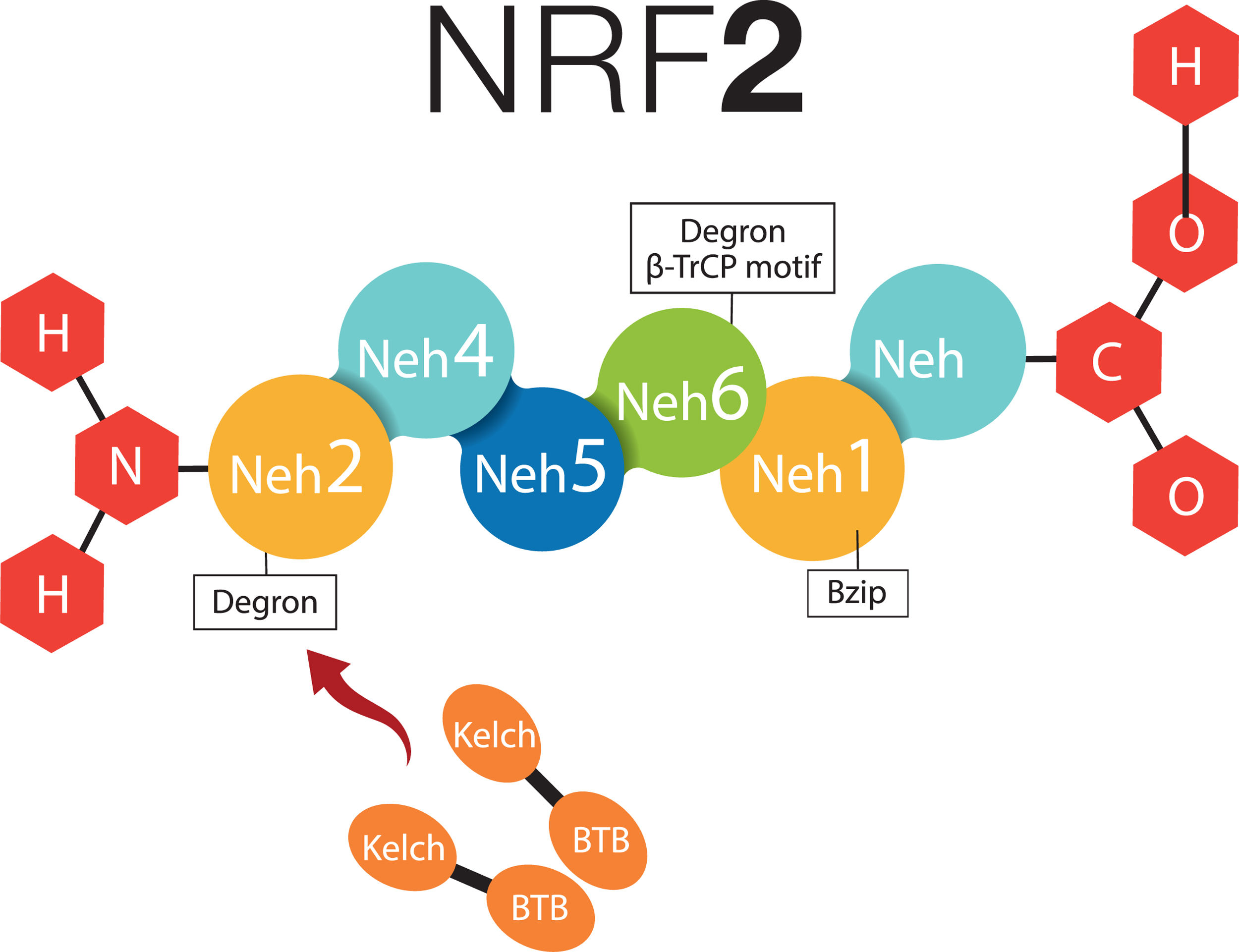
2What about NRF-2 and non-alcoholic fatty liver disease?2.1Cytosolic mechanism of NRF-2Nuclear factor (erythroid-derived 2)-like 2 plays a transcendental role in the antioxidant and anti-inflammatory mechanism, it activates the elements of the antioxidant response (ARE) [9], all this related to Kelch-like ECH-associated protein – nuclear factor (erythroid-derived 2)-like 2 (KEAP1-NRF2) [7,16]. NRF-2 possesses six Neh (NRF2-ECH homology) domains, in which Neh1 is a bZIP structure that works for the dimerization of NRF-2 and its binding to DNA [17]. Neh2 and Neh6 are known as degrons; thus, through β-TrCP (β-transducin repeats-containing proteins), and mainly by means of KEAP1; they are points that can be recognized by the proteasomal degradation systems [18,19]. NRF-2 can normally be found in the cytoplasm, bound to the cytoskeleton, and attached to other proteic complexes responsible for its regulation, principally KEAP1. Therefore, on being exposed to ROS, KEAP1 is oxidized in cysteine and releases NFR-2; at this moment, NFR-2 translocates to the nucleus, in order to dimerize and bind to its corresponding promotor regions to regulate the transcription of the antioxidant response element [20,21]. Neh4 and Neh5 work together for NRF-2-dependent transactivation [19,22] (Fig. 1).
-like 2.")
NRF-2 molecule interacting with KEAP1. NRF-2 has six Neh domains. Neh1 is a Bzip structure that works for the dimerization of NRF-2 and its binding to DNA. Neh2 and Neh6 are known as degrons, which function asregulators in the protein degradation rate. Neh4 and Neh5 together work for NRF-2-dependent transactivation. BTB, BTB domain of KEAP1 protein; Bzip, Basic Leucine Zipper Domain; β-TrCP, β-transducin repeats-containing proteins; Neh, NRF-2-ECH homology; NRF-2, Nuclear factor (erythroid-derived 2)-like 2.
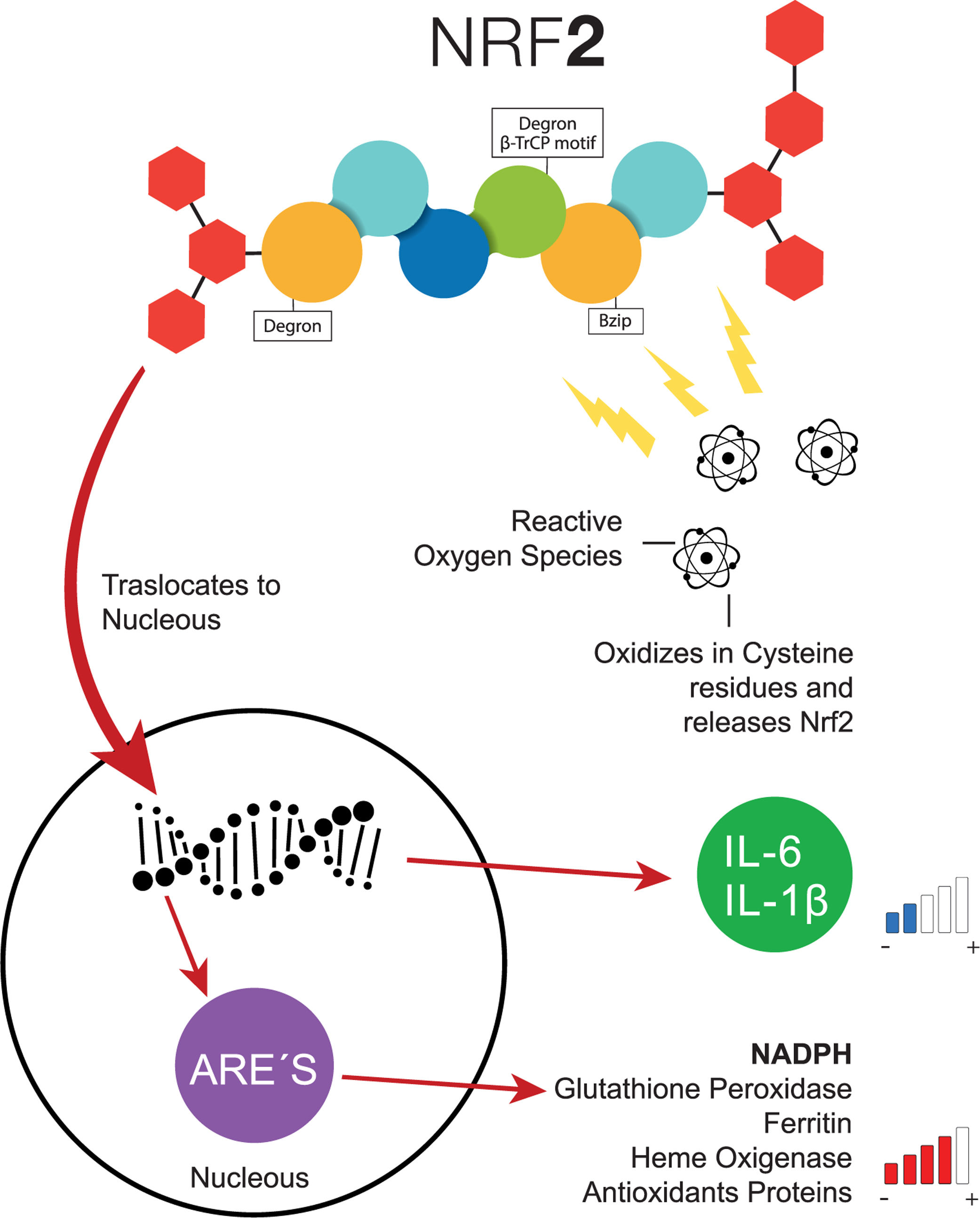
Transcription factor NFR-2 is negatively regulated, principally by KEAP1, via the polyubiquitin pathway by proteasomal degradation. This takes place after the transduction of the messenger RNA (mRNA) from NRF-2, binding a homodimer of KEAP1, and afterward, binding with a full dimer, resulting in the degradation of NRF-2 by the previously mentioned Neh [21]. Nonetheless, when a cell (e.g., a hepatocyte) is exposed to inflammation or oxidative stress, NRF-2 has the capacity to escape from regulation by KEAP1 through the increase in the transcription of mRNA from NRF-2, with the objective of depleting the reserves of KEAP1, which cannot be recycled because of the suppression of another NRF-2 [21]. Therefore, since the NRF-2 protein is not exposed to KEAP1, it can adopt a translocation to the nucleus and join genetic regions that have detoxifying enzymes as an end product (e.g., NADPH, glutathione peroxidase, ferritin, and heme oxygenase-1) or antioxidant proteins [16,22–24]. Additionally, the increase of NRF-2 suppresses the transcription of IL-6 and IL-1β [25]. In addition to KEAP1, other forms of regulatory proteins of NRF-2 have recently been discovered, such as the E3-Ubiquitin-ligase adaptor, and the β-transducin repeats-containing protein (β-TrCP), which can also be degraded by Glycogen Synthase Kinase 3 (GSK-3), phosphorylizing NRF-2 into its residue as serine in the Neh 6 domain, corresponding to the recognition of β-TrCP [18] (Fig. 2).
-like 2.")
Common pathway of NRF-2. When the cells are exposed to oxidative stress, NRF-2 escapes the negative regulation of KEAP1 through the increase of NRF-2, the transcription of interleukins decreases the levels of antioxidant response elements increase. ARE, Antioxidant Response Elements; Bzip, Basic Leucine Zipper Domain; β-TrCP, β-transducin repeats-containing proteins; IL-6, Interleukin 6; IL-1β, Interleukin 1β; NADPH, nicotinamide adenine dinucleotide phosphate hydrogen; NRF-2, Nuclear factor (erythroid-derived 2)-like 2.
The immune system is involved in multiple mechanisms of protection in infectious, neoplastic, poisonous, and other all of which are proinflammatory processes that must be adequately counter-regulated to avoid a persistent or an exaggerated inflammatory process. NRF-2 responds to inflammatory-cell and tissular stimuli with pleiotropic effects that are not yet well defined. The immunological system is also a regulator of lipid deposits through macrolipophagy, where there is phagocytosis of the endo- and exogenous lipids at the hepatic level [26].
In an experimental study conducted by Thimmulappa et al., 2006, the authors reported proinflammatory stimulation with a rise of the level of TNF-α and the induction of sepsis in mice with a deficiency of NFR-2 [27]. In the regulation of innate immunity, the lack of the expression of NRF-2 entails a disturbance in the protective process of the host, as well as increased damage by hyperoxia, the increase of ROS, and inefficiency in the elimination and increase in the production of proinflammatory cytokines [28,29]. The rise in the expression of NRF-2 stimulates the production of Heme oxygenase 1 (HO-1) in different types of cells. The expression of HO-1 through NRF-2 depends on the bioavailability of free heme groups, which are toxic for the cells; these could de-repress NRF-2 and can bind to the gene of HO-1, with the initiation of its transcription [30,31]. HO-1 is in charge of the oxidation of the heme groups free of ferrous iron (Fe2+), carbon monoxide, and biliverdin, the latter being reduced to bilirubin by biliverdin reductase, which acts as a fat-soluble antioxidant [32]. Later, the ROS are selected by other groups derived from the stimulation of NRF-2 as ferritin. HO-1 has antioxidant properties in addition to the anti-inflammatory actions in different tissues [33–35], derivatives of the reduced metabolites of the free heme groups, mainly by pattern recognition receptors (PRR) such as TLR-4 [36]. Glutathione (GSH) is the principal component of the oxide reduction mechanism [37]. GSH and thioredoxin-dependent enzymes (TRX) (i.e., GSH peroxidase, GSH reductase, GST, and TRX reductase) are the main antioxidant systems controlled by NRF-2. GSH and TRX can be regenerated by GSH reductase and TRX reductase, respectively [37]. The absence of GSH peroxidase with the concomitant decrease of GSH leads to atherosclerosis associated with diabetes and gastrointestinal inflammation [38,39].
The qualitative or quantitative dysfunction of the regulatory T cells (Treg) leads to a series of autoimmune diseases with more established mechanisms [40]; in addition, Treg are suppressed by the action of the proinflammatory cytokines secreted by other inflammatory cells [40]. During an experimental study, when NFR-2 was activated in Treg, the inflammatory action was decreased due to the rise in Treg cellular levels [41] and, in another experimental method, it was proven that the anti-inflammatory mechanism is also found regardless of the presence of Treg [16] With synthetic antioxidants, NRF-2 can be activated in T cells determined by the rise in the antioxidant response elements mediated by NRF-2, as well as the suppression of IFN-γ and the concomitant production of the cytokines of Th2 lymphocytes, such as IL-4, IL-5, and IL-13; thus, the functional increase of NRF-2 entails a rise in the activation of Th2 lymphocytes [42]. Also, it significantly increases the half-life of the mRNA in IL-8 through its stabilization and the expression of the proinflammatory cytokines, which contain ARE-like elements in their promotor region [43,108].
2.3Nonalcoholic fatty liver disease and NRF-2To define nonalcoholic fatty liver disease, it should be proven, by means of an image or a histological method, that the steatosis is not related to any secondary cause that conditioned the accumulation of liver fat, such as alcohol consumption, infection by hepatitis C virus, or any congenital metabolic disorder, steatogenic medications or among other causes. Nonalcoholic fatty liver disease is one of the most prevalent hepatic diseases worldwide, with different percentages of prevalence related to the group studied, and with associated risk factors or comorbidities [2]. In the majority of patients, Nonalcoholic fatty liver disease (NAFLD) It is associated with metabolic diseases, such as diabetes mellitus, obesity, or dyslipidemia [7]. Liver injury in NAFLD is caused by two mechanisms in general: (1) by peripheral resistance to insulin, and (2) by intrahepatic oxidative stress, whether with an increase of reactive oxygen species and reactive nitrogen species (ROS and RNS) and/or as a result of the decrease in antioxidant tissue levels. The increase in ROS or RNS levels involves a concomitant increase of proinflammatory cytokines and the activation of NF-κβ, which in turn activates the hepatic stellate cells, the subsequent fibrosis, and damage to the DNA [12,64], with a failure in the synthesis of exogenous antioxidants [12]. Physiopathological consequences with clinical expression have been observed in different systems, such as that recently found by Chang-Hoon et al., in which patients with fatty liver had lower lung function as measured by FVC [45]. The youngest population is severely affected, with insulin resistance the common proinflammatory condition [5]. The body mass index (BMI) has a significant correlation with the increase of TNF-α and C-reactive protein [4]. Patients with NAFLD have high levels of insulin and circulating leptin, giving rise to the activation of renal mesangial cells and tubular inflammation through the COX-2 (Cyclooxygenase type 2) pathway [46]. Type 2 diabetes is considered an independent risk factor for progression and severe fibrosis in patients with fatty liver [47]. It has been observed that patients with fatty liver and type 2 diabetes register histological data of greater severity than patients without diabetes [7]. It is classified based on histological findings with hepatic steatosis when it only has a fatty accumulation, and steatohepatitis is defined with histological findings of inflammatory tissue [2]. Hepatic fibrosis in patients with NASH have a mean annual progression rate of 0.09% (95% CI: 0.06–0.12) [3].
The liver is a vital organ in which numerous metabolic functions take place; therefore. it is exposed to ROS, primarily in the mitochondria and systems dependent on P450 cytochromes, related principally to lipid synthesis, degradation, the oxidation of fatty acids, and the metabolism of cholesterol and phospholipids for diverse cellular functions, including structural, transport, and others [48,49]. Hypoxia-inducible factors 1α and 2α (HIF-1α and HIF-2α) exert different effects on lipid metabolism [50]. The overexpression of HIF-2α promotes fatty liver due to the diminution of the β-oxidation of fatty acids and induces lipogenesis through the PPARα pathway [50].
In an experimental study carried out by Chen Y et al., 2018, it was determined that, in Sprague Dawley rats fed a High fat diet (HFD) during 12 weeks, the authors demonstrated that APS1 significantly reduced body weight and also reduced the accumulation of hepatic lipids regulating the expression of SIRT-1/PGC-1α/SREBP-1, as well as the increase in hepatic antioxidant activity on modulating the expression of activity of NRF-2 and HO-1 [51]. In experimental models with hepatic steatosis induced by a fat-rich diet, a diminution was found of transcription due to the diminished levels of mRNA of the components AMPK-PGC-1α, NRF-2, and β-ATP synthase [52].
Recent experimental and a few clinical research findings have found sufficient physiopathological evidence that proves that an excess of energy reserves in humans with problems such as nonalcoholic fatty liver disease or diabetes is harmful. Thus, the excess of glucose and other carbohydrates such as fructose can be transformed into lipids that can be stored as triglycerides; therefore, a higher quantity of fatty acids requires a higher amount of β-oxidation and a subsequent rise of free radicals [53,54]. An excess in the quantity of fructose levels in the bloodstream has been demonstrated, by clinical and experimental investigations, to increase lipogenesis. The oxidation of fatty acids is affected as a result of the metabolism of fructose by fructokinase C, thus the result of the consumption of adenosin triphosphate (ATP), the production of uric acid and the nucleotide exchange, and finally, concomitant mitochondrial dysfunction [55]. RIP3 increases according to lipid accumulation and concomitantly the lipogenesis and signaling of NF-κβ, in addition to the inhibition of the NLRP3 imflammasome [56]. By means of experimental models, it was found that in rat, RIP3−/− (receptor-interacting kinase-3) diminishes inflammation, steatosis, and oxidative stress through the previously mentioned mechanisms and through the greater activation of NRF-2/HO-1 [56]. The Unfolded Protein Responses (UPR) are critical steps in cellular stress and inflammation in patients with NASH [13,109]. The signaling network of the mitochondrial Unfolded Protein Response (UPRmt) and mitohormesis are a retrograde signaling pathway with communication from the mitochondria to the nucleus. These factors inhibit the progression of fatty liver and hepatic fibrosis, as well as the disminution of resistance to insulin [57,109]. According to Puri et al. in 2008, patients with NASH possess failure to mobilize an XBP1-mediated increase in splicing in EDEM1, which promotes the proteomic degradation of ubiquitinated proteins. This mechanism additionally is accompanied by the phosphorylation of JNK1, as a result affecting insulin signaling, giving rise to an increase in inflammation and in apoptosis [58]. The aberrant expression of circScd1 affects hepatocellular lipidosis and promotes fatty liver through the JAK2/STAT5 pathway [59]. The growth hormone regulates several vital processes at the systemic level and at the hepatic level, the latter through the JAK2/STAT5 pathway [60]. This JAK2/STAT5 pathway possesses ambivalent functions in the liver; thus, it is observed as implicated in the development of cancer and of hepatic stenosis [60]. The group of Gregory Gore described cell death through the mechanism of apoptosis in humans with humans with NASH and successfully introduced the definition of lipotoxicity in patients with NASH [61]. The progression from hepatic steatosis to steatohepatitis and cirrhosis is closely related to OS, lipotoxicity, and local inflammation [62,63]. Likewise, the elevated levels of ferritin activate proinflammatory signaling through NF-κβ with a later increase of interleukins, principally IL-1β [5]. Glutathione S transferase (GST) is the main intracellular antioxidant synthetized by Glutamate-Cysteine ligase and glutathione synthase, as demonstrated by Chen et al., 2007 in a study including with Gclc Knockout mice, which developed steatosis in very early ages [65]. The most important transcription factor in the regulation of antioxidant enzymes is NRF-2 [66]; hence, it controls genes that contain ARE, electrophilic-conjugating enzymes, ubiquitin/proteasomes, and heat-shock proteins, in response to reactive oxygen species [67]. Hepatocytes possess highly efficient counter-regulatory systems against oxidative effects such as cytochrome systems (CYP) P450, either by endogenous or exogenous substances, enzymes such as glutathione S transferase, the flow-through transporters, and molecules that contain “thiol”, such as Glutathione or Thioredoxin [68]. Many of the antioxidant mechanisms are encoded by genes that contain ARE (antioxidant response elements), which are activated by the increase of free radicals [69]. The activation of NRF-2 in liver suppresses the synthesis of fatty acids [70], as observed in this study that included experimental feeding with 8-week-old NRF-2 null mice, and wild-type mice fed experimentally, observing the mRNA of sterol regulatory element-binding protein 1-c and fatty acid synthase as highly expressed in NRF-2 null [71], concluding that the protective action is afforded by the increase of AR. The findings are noteworthy of Qiao Y. et al., in which the authors reported the role of inducible nitric oxide synthase (iNOS) in the pathogenesis of steatohepatitis, with a protector role on increasing the expression of Heme oxygenase-1 (HO-1), similarly regulated by NRF-2 [44]. Oxidative stress is a fundamental step for the development of steatohepatitis, experimentally demonstrated in the liver of knockout mice with NFR-2 −−/−− and with a deficient diet of methionine and choline for 14 days, which had a significant increase of neutrophils in the tissue, micro steatosis, and macrovascular steatosis compared with wild-type mice, and a 10-fold increase of KF-κβ and p65 and a 5-fold increase of IL-1β, mRNA, and TNF-α. Therefore, the qualitative and/or quantitative deficiency of NRF-2 comprises a fundamental mechanism in the development of steatohepatitis [73]. CYP2E1 and CYP4A are known to be linked with the physiopathological development of steatohepatitis [74]. CYP2A6 is the phase I regulator of a great amount of endogenous and exogenous molecules [75], and its elevation is related to the development of liver pathologies [76]. The homologous cytochrome CYP2A6 of humans in Cyp2a5 mice is expressed in several liver diseases by the induction of toxic chemicals [77], where the levels of mRNA and Cyp2a5, on comparison between knockout NRF-2−/− and wild-type mice, proved the increase of NRF-2 and Cyp2a5 in wild-type mice, as well as their interaction by co-immunoprecipitation Cyp controlled by NRF-2 [77]. With these results, the manner was once again proven in which hepatic stenosis affects the homeostasis of NRF-2.
2.4Therapeutic advances of NRF-2 and nonalcoholic fatty liver diseaseIn 2016, the “IQ DILI Initiative” was created, comprising a scientific group focused on the prevention of DILI in the development and investigation of new drugs; the group was composed of 39 pharmaceutical and biotechnology companies [78]. There is no clear evidence that patients with Chronic liver disease (CLD) have a greater incidence of Drug-induced liver injury (DILI); however, it has always been accepted that patients with an underlying hepatic disease is predisposed to drug-induced liver injury and that the latter will be more severe [79]. Patients with NAFLD exhibit an increase in the activity of CYP2E1 and a diminution of 1.9 and 3.1 times in the activity of CYP3A4 in NAFLD and Non-alcoholic Steatohepatitis (NASH), respectively [80,81]. Similarly, diabetes is a factor of diminution of the activity of cytochrome CYP3A4 [81].
The main risk factors related to the development of fatty liver disease are obesity and type 2 diabetes, but what is it that occurs between type 2 diabetes and transcription factor NRF-2? In an experiment conducted by Petersen et al., 2018, the authors demonstrated that, in mice fed a standard diet but with the elimination of liver-specific phosphatase and the tensin homology (PTEN) (PTENLKO), the animals had more steatosis and hepatic fibrosis compared with the controls. These authors concluded that organisms with deficient PTEN had more injuries due to OS and, as a result, greater steatohepatitis and fibrosis [82].
According to the guidelines established for the management of patients with nonalcoholic fatty liver, to date the drugs that have been investigated in clinical assays, such as Pioglitazone, have histological benefits in patients with NASH with and without diabetes [2]. With the GLP-1-group drugs, nowadays there is insufficient evidence for their recommendation for the treatment of patients with steatohepatitis [2]. Improvements have been observed at the histological level with vitamin E in patients without diabetes, but there is no evidence proving its benefit in patients with diabetes. Ursodeoxycholic acid and omega-3 fatty acids are not recommended for the treatment of NASH [7]. For their part, type 2 Sodium-glucose cotransporter inhibitors (iSGLT2), by means of experimental assays in animals [83,84] and clinical studies, have demonstrated the diminution of oxidative stress and, at the hepatic level. with clinical, biochemical, and imaging parameters [85–89], due to the diminution of cytokeratins 18-M30 and 18 M-65 [85] and the diminution of type 7S collagen [86]. On the other hand, there are studies in which the results have not favored the use of iSGLT2 for the treatment of non-alcoholic fatty liver, in that there was no diminution in the FIB-4 fibrosis scale and in the NAFLD score [90].
Experimental investigations from cellular cultures found the activation of NRF-2 by high doses of glucose, but there was variability between type of cellular pedigree and time of exposure [91–93]. The inducing components of NRF-2 have been classified into the following four groups: 1. phenolic antioxidants; 2. dithiolethiones; 3. isothiocyanates, and 4. triterpenoids [70,94].
According to Yoh et al. in 2008, the dysregulation of NRF-2 is necessary in the development of type 2 diabetes and its complications [95]. To date, there is no proven and approved treatment for clinical use in patients with nonalcoholic fatty liver disease with significant efficacy; however, a clinical trial was conducted in which Liraglutide was administered to a group of rats and a placebo to rats in the other group; the authors concluded that the group exposed to the drug had an increase in the expression of NRF-2, as well as ion the blood and hepatic levels of GSH (p<0.05) [96].
There are several theories of anti-inflammatory components in the NRF-2 pathway that, in a selective manner, intervene in the inflammatory process at pulmonary, renal, urinary, and hepatic levels [19]. The antioxidant properties of several natural compounds related to the activation of NRF-2 have been investigated [43], such as curcumin [97], KIOM-4, Resveratrol, Zinc, Berberine, L-arginine, oleanolic acid, vitamin D, among others, in animal cell cultures, with disagreeing results, mainly focused on the decrease of OS or the increase of elements of antioxidant response through NRF-2 [43]. Macías-Pérez et al. in 2019 published a study that demonstrated that curcumin increases the expression of the mRNA of NRF-2 and of NRF-2/NF-κβ [97]. There have been investigations of drugs (e.g., Silymarin) in which its mechanism of action is the activation of NRF-2 either directly or indirectly, and the decrease of the levels of OS components, specifically in liver diseases such as fatty liver [98,99]. In an experimental study conducted by Ghowsi et al., 2018, the authors evaluated OS in the liver and blood of mice, proving that the levels of malondialdehyde and the homeostatic model assessment of insulin resistance (HOMA-IR) were higher in the control group of mice compared to the group with Resveratrol (p=0.034 and p=0.014, respectively) [100]. The benefits of Resveratrol had been proven through diverse models, as in diabetic mice along the NRF-2/HO-1 pathway [101], as well as a better glucose and lipid profile through the activation of Keap-1/NRF-2 [102]. In some herbs, great quantities have been found of components that activate NRF-2, increasing the expression of cytoprotector genes. The most cited examples include curcumin extracted from ginger, isothiocyanates retrieved in broccoli, and anthocyanins from berries and grapes [19,103]. Raphanus sativus L. var. niger, which is well known as black radish, exerts a hepatoprotector effect mediated by NRF-2/HO-1 when the liver is exposed to Carbon tetrachloride (chemical substance with a hepatotoxic effect) [104]. Likewise, the pharmacological option has been investigated with the negative regulation of NRF-2, mainly with cytotoxic agents, as in acute monocytic leukemia [105]. A study was conducted in mice exposed to excessive fat diets and 1,25 hydroxycholecalciferol, then the determination of lipids resulted in decreased levels of lipids in the blood and intrahepatic lipids, and also attenuated the hepatic steatosis and inflammation, as well as MDA and F2 α-isoprostane markers of intrahepatic oxidative stress [106]. NRF-2 translocated to the nucleus, not by direct regulation of mRNA, and raised the expression of genes that encode antioxidant enzymes [106].
There is research being conducted currently with modified supplements that induce the activation of NRF-2 through several mechanisms known to have achieved experimental success, but without clinical evidence to support its use [107]. All this information that is being generated on the molecular and pharmacodynamic mechanisms leads us to see the need to create multidisciplinary investigative groups for the development of hepatoprotectors or hepatic antioxidants. In view of the new technologies in the investigation and development of pharmacology, it is necessary to advance in the therapeutics of these patients with fatty liver, mainly without diabetes, due to that, according to Cho et al. (2019), administering treatment to patients with fatty liver without diabetes diminishes the risk of suffering from diabetes and its probable consequences [72].
3ConclusionThe complex interaction between various cytosolic and intranuclear components with NRF-2, although better understood, continues to encounter a lack of information, leading us to clearly identify under which conditions its action is activated or suppressed by means of various, already identified clinical situations. The role of NRF-2 has been researched in recent years, as a protector against the oxidant and inflammatory mechanism and its place in various diseases as principal agent. Nonalcoholic fatty liver disease has become one of the hepatic diseases of greatest interest recently due to its epidemiological impact, with impressive clinical and physiopathologic advances and more profound knowledge about its mechanisms that lead to persistent fibrosis, which is intimately related with NRF-2.
Multiple drugs have been tested with an antioxidant and anti-inflammatory objective, through the NRF-2 pathway, without being approved as possessing evident clinical benefit to date. While the role of NRF-2 has been proven in hepatic pathogenesis and it has been established as a potential therapeutic target, it is at present under worldwide researches.AbbreviationsAMPK-PGC-1α Activated protein kinase coactivator 1 alpha antioxidant response element chronic liver disease cytochromes glutathione S transferase glutathione high fat diet heme oxygenase 1 inducible form of nitric-oxid Synthase janus kinasa 2 Kelch-like ECH associating protein-1 liver specific phosphatase and tensin homology mitochondrial unfolded protein Response nonalcoholic fatty liver disease nonalcoholic steatohepatitis nuclear factor (erythroid-derived 2)-like 2 peroxisome proliferator-activated PTEN deletion mice reactive nitrogen species reactive oxygen species signal transducer and activator of transcription 5 sirtuine 1 sterol regulatory element-binding protein-1 thioredoxin-dependent enzymes unfolded protein response
The authors have no conflicts of interest to declare whatsoever.



