



HBV covalently closed circular (ccc) DNA is the key player in viral persistence and an important predictive biomarker for hepatitis relapse. Precise quantification of intracellular cccDNA is challenging because cccDNA is present in very low levels in hepatocytes, where it also co-exists with a large excess amount of relaxed circular (rc) DNA. We aimed to develop a highly sensitive cccDNA detection method for cccDNA quantification by digital PCR (dPCR).
Patients or materials and methodsA standard plasmid containing the whole HBV genome in the closed circular conformation was employed to characterize the performance of dPCR. rcDNA in the growth medium of HBV-producing HepAD38 cells was used as a matrix for cccDNA detection. Intrahepatic cccDNA measurement by dPCR and qPCR was performed to determine the correlation of the analysis results for the two methods.
ResultsThe limit of detection (LOD) of the cccDNA dPCR was 1.05copy/μl, and the linear range of detection was 1.02×104copies/μl, achieving a dynamic detection range of 104-fold. cccDNA measurement using excess rcDNA as the matrix did not reveal false-positive detection, indicating that dPCR was highly specific. In the HepAD38 cells, the cccDNA levels measured by dPCR were highly correlated with those measured by qPCR but had a higher sensitivity. The CDK inhibitor AZD-5438 was found to block intracellular cccDNA synthesis.
ConclusionsDpcr greatly improved the sensitivity and specificity of cccDNA detection. Host CDK activities are likely required for cccDNA synthesis. dPCR can potentially be applied for drug screening for effective cccDNA inhibitors.
Chronic hepatitis B (CHB) virus infection is the most important cause of hepatocellular carcinoma (HCC) worldwide [1,2]. Currently, more than 2 billion people have been infected with HBV, and 350 million among them suffer from CHB, which often results in cirrhosis and HCC, diseases that are among the top leading causes of death worldwide [3–7]. In some countries, HBV vaccination for newborns has been launched to reduce infection rates [8]. Additionally, antiviral therapies using nucleotide/nucleoside analogs have achieved effective “functional” cure of the virus infection. Despite this valuable progress in vaccines and treatments, so far there are still no reliable strategies for “complete” or “virological” cure due to the difficulty of eradicating the HBV residual replication intermediate covalently closed circular (ccc) DNA in hepatocytes [9]. Therefore, developing high-risk biomarkers for viral reactivation after nucleotide/nucleoside (NA) therapies in CHB patients is a very important issue.
HBV enters the cell by binding to the sodium taurocholate cotransporting polypeptide (NTCP) receptor on the surface of hepatocytes [10,11]. After entry, the viral nucleocapsid is released into the cytoplasm and then translocated into the nucleus [10]. Within the nucleus, relaxed circular (rc) DNA is converted into cccDNA, which then serves as a template for transcription [12]. The cccDNA functions as the template for transcription of the four viral RNAs (0.7, 2.1, 2.4, and 3.5kb), which are exported to the cytoplasm and used as mRNAs for translation of the HBV proteins. The longest (i.e., pregenomic, pg) RNA functions as the template for replication mediated by the viral polymerase, which executes the action of reverse transcription. Among all the steps in the HBV life cycle, reverse transcription is currently the main target for antiviral therapies. Nucleotide/nucleoside analogs, such as adefovir and entecavir, which block the process of viral reverse transcription, have been widely used as treatments for CHB patients. The drugs in this category are generally well tolerated, and potent drugs, such as entecavir and tenofovir, can often reduce viremia to below the detection limits of standard viral DNA measurement methods. However, viral reactivation after therapy discontinuation often occurs (∼60% within 2 years post treatment cessation), suggesting that lifelong treatment is necessary [13].
Because nucleotide/nucleoside drugs inhibit viral replication in the relative “late” phase of the viral life cycle and do not block the replication products generated in earlier phases, recent efforts to develop new strategies in HBV therapy target various critical steps involving in HBV replication and propagation, including viral entry, encapsidation, gene expression, and virion assembly. These novel antiviral drugs provide unprecedented opportunities to destroy HBV cccDNA and other replication intermediates and completely eradicate the virus, but the main challenge yet unresolved is the reliable quantification of cccDNA in the liver in the course of drug treatments [14]. Monitoring the ability of these treatments to reduce the cccDNA level, which is the key replication reservoir, is an essential task to demonstrate their potential to reach virological cure.
The molecular mechanism of HBV cccDNA synthesis remains obscure despite of recent identification of the HBV receptor, NTCP, which has made de novo infection experiments in cultured hepatocytes much more applicable [15]. rcDNA, which is reverse transcribed from pgRNA, contains the phosphotyrosyl-bonded polymerase (P) protein and a single-stranded flap, which is believed to be removed by host endonucleases. Conversion of partially double-stranded and nick-containing rcDNA to fully double-stranded cccDNA predictably requires the actions of a DNA polymerase, a kinase, endonucleases, a ligase, a topoisomerase, and DNA repair factors. Additionally, it was reported that the phosphotyrosyl-linked P protein can be resolved by specific tyrosyl-DNA-phosphodiesterases (TDPs). However, many important players involved in the process of cccDNA synthesis remain to be characterized [16].
Some recent studies have reported the potential role of the host cell cycle machinery in HBV replication and propagation. The cyclin E2-cyclin-dependent kinase (CDK) 2 complex has been reported to be involved in regulating viral replication through mediating phosphorylation of the SAM domain- and HD domain-containing protein 1 (SAMHD1) [17]. Additionally, HBx, a protein important for HBV cccDNA synthesis and carcinogenic processes, has been found to activate protein kinase C (PKC), a family of protein kinases that are involved in controlling the function of some CDK proteins through phosphorylating serine and threonine amino acid residues on these proteins, leading to cell cycle progression [18]. PKC has also been shown to facilitate pgRNA encapsidation and virion production [19]. In light of these previous findings, we hypothesized that manipulation of the host cell cycle progression pathway represents a good candidate to control cccDNA synthesis.
As the main viral replication reservoir, HBV cccDNA plays a key role in HBV persistence [16]. Therefore, cccDNA is believed to be an important predictive biomarker for the risk of adverse CHB outcomes. However, reliable detection of cccDNA with high sensitivity and specificity has been challenging because the cccDNA level is very low in the cell (2–30copies/cell), and it co-exists with a large excess amount of rcDNA [20]. The recent revolutionary digital PCR technology provides quantification of target nucleic acids with high sensitivity and specificity. We therefore hypothesized that digital PCR is a promising approach for cccDNA detection in antiviral drug development [21]. In this study, we established a convenient and cost-effective platform for the sensitive detection of cccDNA and explored the host molecular pathways controlling cccDNA synthesis.
2Materials and methods2.1Cell lineHepAD38 cells, which contain the HBV genome under the tet-off transcription control, were used to examine the intracellular amplification of the virus genome and propagation [22]. The cells were maintained at 37°C in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS), 200mM L-glutamine (Gibco, Life Technologies, Carlsbad, CA, USA), 10U/μl penicillin-streptomycin (Lonza, Basel, Switzerland), and 400μg/ml G418 (MDBio) with 5% carbon dioxide. The cells were also maintained in medium supplemented with 0.3μg/ml doxycycline to keep viral gene transcription shut off until the time for viral gene expression.
2.2Propagation of HBVTo induce viral gene expression and propagation, HepAD38 cells were grown in doxycycline-free medium. Virions were harvested every 5 days from the growth medium of the cultured cells and then filtered through a 0.45-μm filter. Viruses in the filtrate were concentrated using 10% polyethylene glycol (PEG) 8000 with gentle rotation at 4°C overnight, followed by centrifugation at 7000g for 0.5h. The supernatant was discarded, and the precipitated viruses were re-suspended in 2ml of serum-free DMEM/F-12 medium and then stored at −80°C. The harvested viral DNA was quantified by real-time PCR (Roche).
2.3HBV DNA extractionHBV DNA was extracted from HepAD38 cells that were grown in medium containing no doxycycline to propagate HBV. After harvest, the cells were treated with 0.05g/L RNase A at 37°C for 30min, followed by cell lysis buffer (0.1% SDS, 150mM NaCl, 20mM EDTA, 20mM Tris-HCl [pH 7.5], and 0.8g/L proteinase K) at 55°C overnight. DNA in the cell lysate was then extracted with equal volumes of phenol and chloroform twice and chloroform once. DNA in the aqueous phase was mixed with 2 volumes of isopropanol and then centrifuged for precipitation.
2.4HBV cccDNA extractionHBV cccDNA was isolated from HepAD38 cells that were grown in medium containing no doxycycline. The cells were lysed with the modified Hirt solution (1% SDS, 50mM Tris–HCl [pH 8.0]), 10mM EDTA [pH 8.0], and 150mM NaCl) [23]. After 30min of incubation at room temperature, 1/5 volume of 5M NaCl was added to the cell lysate and incubated at 4°C overnight. After centrifugation, the supernatant was supplemented with proteinase K (50μg/ml) and incubated at 37°C to digest protein for 2h. cccDNA was purified by phenol and chloroform extraction and ethanol precipitation. After centrifugation, the pellet was allowed to air dry and was dissolved in ddH2O. The purified cccDNA was treated with T5 and T7 exonucleases (New England Biolab) to digest contaminating rcDNA.
2.5HBV rcDNA extraction from cell culture mediumHBV DNA in the cell culture medium was extracted using a QIAamp MinElute Virus Spin Kit (Qiagen). Briefly, doxycycline (-) culture medium of virus-producing HepAD38 cells was incubated with proteinase K and Buffer AL, which contained carrier RNA to facilitate extraction, then incubated at 56°C for 15min. The treated DNA was mixed with ethanol, incubated at room temperature for 5min and then precipitated by centrifugation. DNA was resuspended then loaded into a QIAamp MinElute column, and washed with Buffer AW1 and AW2 by gravity. DNA in the column was eluted with RNase-free ddH2O.
2.6Cell survival assayHepAD38 cells were treated with various doses of the chemical UCN-01 (Sigma-Aldrich) or the solvent DMSO for 48h. The cell survival rates were detected by the (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tetrazolium (MTT) assay. Briefly, the cells were incubated in serum-free DMEM containing 0.5mg/ml MTT (Sigma–Aldrich) at 37°C for 3h. After the reaction, the MTT solution was transferred to a 96-well ELISA plate, and the absorbance was measured at 570nm.
2.7HBV cccDNA quantitative PCRVirus-producing HepAD38 cells were treated with various concentrations of UCN-01 (Sigma-Aldrich), the PKC inhibitor Go 6983 (Sigma-Aldrich), or the CDK inhibitor AZD-5438 (Sigma-Aldrich) for 48h before harvest. The cccDNA extracted with the Hirt solution was treated with the T5 and T7 exonucleases to remove rcDNA. One microgram of cccDNA was treated with 5U each of the T5 and T7 exonucleases at 30°C for 90min, followed by incubation at 99°C for 5min to stop the enzymatic reactions. After digestion, cccDNA quantitative PCR was performed following a previously described protocol [24]. Briefly, the cccDNA template was mixed with TaqMan Universal Master Mix II and TaqMan® Probe (Thermo) for cccDNA detection and then subjected to the real-time PCR reactions. The PCR program was 50°C for 2min, 95°C for 10min, and then 40 cycles of 95°C for 30s and 60°C for 30s. The nucleotide positions and sequences of the PCR primers and the TaqMan® probe were forward primer: 5′-(1562) TTCTCATCTGCCGGACCG (1579)-3′, reverse primer: 5′-(1883) CACAGCTTGGAGGCTTGAAC (1864)-3′, and probe: 5′-FAM-(1836) CCTAATCATCTCTTGTTCAT (1855)-MGB-3′. The cccDNA-specific PCR primers span the gap region (1611–1838) on rcDNA.
2.8HBV cccDNA digital PCRThe cccDNA was quantified by digital PCR using the Clarity™ Digital PCR System (JN Medsys) following the manufacturer's instructions. The PCR primers and TaqMan® Probe were the same as those used in the cccDNA quantitative PCR described in the previous section. The cccDNA template was partitioned into a multi-well PCR chip, where the PCR was divided into thousands or more of nanoliter sub-reactions such that each had at most a single copy of DNA. The PCR program for digital PCR was 95°C for 5min and then for 40 cycles of 95°C for 50s, 55°C for 100s and 70°C for 5min. After the PCR reactions, the images of the PCR chip results were scanned and converted into scatterplots of signals of the HEX dye conjugated to the hybridization probe in the PCR, and the cccDNA copies present in the sample were calculated.
2.9HBV pgRNA quantification by real-time RT-PCRRNA of the HepAD38 cells was extracted using REzol™ C&T buffer (Protech) and then reverse transcribed into cDNA, following previously described protocols [25]. HBV pgRNA was detected by quantitative RT-PCR. Briefly, the cDNA was mixed with Fast SYBR® Green Master Mix (Thermo) and the pgRNA-specific PCR primers, and the reaction was then amplified by real-time PCR (Roche). The sequences of the RT-PCR primers were forward: 5′-TGTGGAGTTACTCTCGTTTTTGC-3′ and reverse: 5′-AAGGCTTCCCGATACAGAGC-3′ [26].
2.10Detection of HBV core and surface proteins by Western blottingHBV core and surface proteins were detected in the virus-producing HepAD38 cells by Western blotting, following a previously described protocol [25]. The antibodies used were an anti-HBsAg antibody (7H11-HRP, generously provided by Prof. Jun Zhang, Xiamen University, China) and an anti-HBcAg antibody (Novocastra).
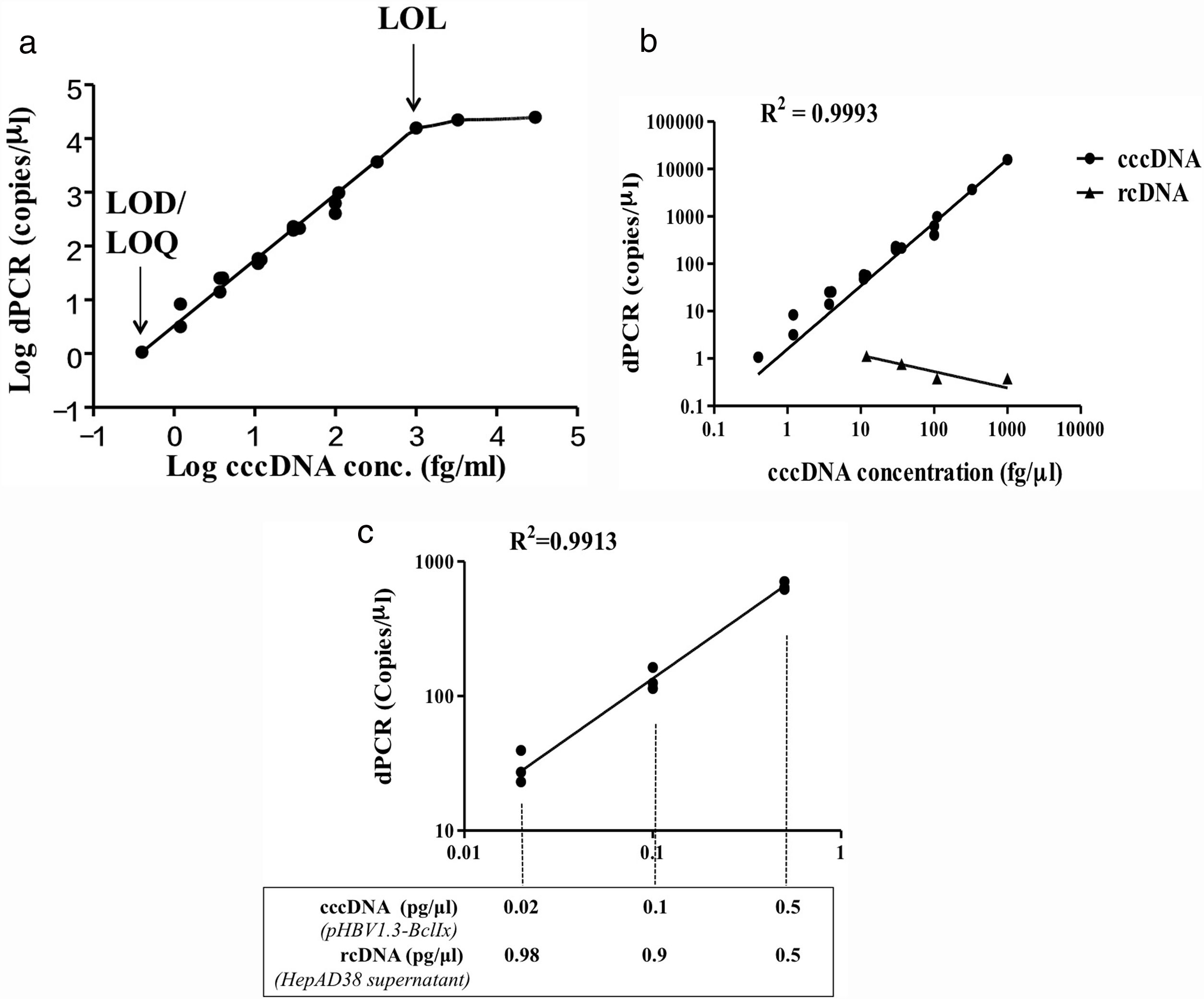
3Results3.1Development of HBV cccDNA analysis by digital PCRWe aimed to establish a digital PCR method to precisely quantify HBV cccDNA. The plasmid pHBV1.3x-BclI, which contains the whole HBV genome in double-stranded closed circular conformation, was used as the positive control for cccDNA and first subjected to the analysis [27]. The results showed that for the pHBV1.3X-BclI plasmid at various concentrations, the DNA copy numbers revealed in the digital PCR analysis were highly consistent with the input of the target HBV plasmid (R2=0.9993) but not rcDNA, showing a high specificity of cccDNA quantification by the ClarityTM dPCR platform (Fig. 1B). Based on multiple independent experimental results, the limit of detection (LOD) and limit of quantitation (LOQ) were 1.05copy/μl, and the limit of linearity (LOL) was 1.02×104copies/μl, indicating that the linear range of detection was 1.05 to 1.02×104copies/μl, reaching a dynamic detection range of 104-fold (Fig. 1A).
 LOD, LOQ, LOL, and linearity range of cccDNA quantification using the HBV plasmid pHBV1.3x-BclI as a standard. Based on at least 3 independent experiments, the LOD and LOQ for HBV cccDNA digital PCR were 1.05copy/μl, and the LOL was 1.02×104copies/μl. The linear range of detection was 1.02×104copies/μl. (B) Quantification of cccDNA in the pHBV1.3-BclI plasmid and rcDNA in virions in the growth medium of virus-producing HepAD38 cells. For the pHBV1.3X-BclI plasmid, the cccDNA copy numbers measured by digital PCR were consistent with the input numbers (R2=0.9993). However, the rcDNA in the same amounts was not detected by cccDNA digital PCR, indicating that digital PCR by the Clarity™ dPCR platform was highly sensitive and specific for cccDNA detection. (C) Quantification of cccDNA in reactions with various amounts of rcDNA. The relative ratios of cccDNA to rcDNA were 1–1, 1–9, or 1–49 in the digital PCRs. The dotted lines indicate the amounts of cccDNA template in the digital PCR reactions. In various ratios of cccDNA to rcDNA, the cccDNA copy numbers can be accurately quantified (R2=0.9913).")
Development of digital PCR for HBV cccDNA quantification. (A) LOD, LOQ, LOL, and linearity range of cccDNA quantification using the HBV plasmid pHBV1.3x-BclI as a standard. Based on at least 3 independent experiments, the LOD and LOQ for HBV cccDNA digital PCR were 1.05copy/μl, and the LOL was 1.02×104copies/μl. The linear range of detection was 1.02×104copies/μl. (B) Quantification of cccDNA in the pHBV1.3-BclI plasmid and rcDNA in virions in the growth medium of virus-producing HepAD38 cells. For the pHBV1.3X-BclI plasmid, the cccDNA copy numbers measured by digital PCR were consistent with the input numbers (R2=0.9993). However, the rcDNA in the same amounts was not detected by cccDNA digital PCR, indicating that digital PCR by the Clarity™ dPCR platform was highly sensitive and specific for cccDNA detection. (C) Quantification of cccDNA in reactions with various amounts of rcDNA. The relative ratios of cccDNA to rcDNA were 1–1, 1–9, or 1–49 in the digital PCRs. The dotted lines indicate the amounts of cccDNA template in the digital PCR reactions. In various ratios of cccDNA to rcDNA, the cccDNA copy numbers can be accurately quantified (R2=0.9913).
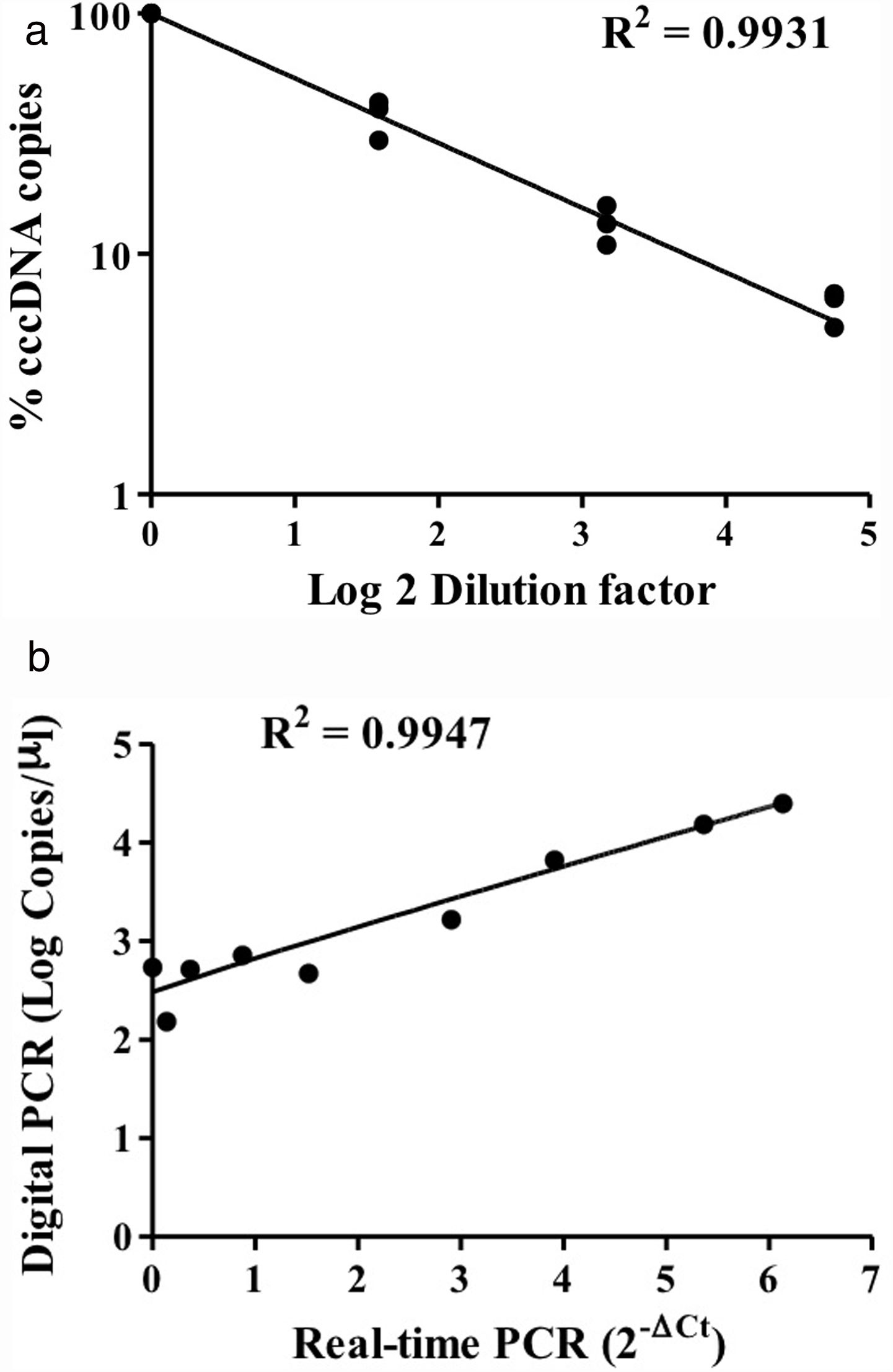
HBV rcDNA, which co-exists with cccDNA in the cell, often interferes with cccDNA detection. To examine the specific detection of cccDNA in the presence of excess rcDNA, we used rcDNA as the matrix for cccDNA detection. The two types of DNA molecules were mixed in various molecular ratios and then subjected to cccDNA digital PCR with the Clarity™ platform. HBV rcDNA was isolated from virions in the growth medium of cultured HBV-producing HepAD38 cells. Samples in the cccDNA to rcDNA ratios of 1–1, 1–9, and 1–49 were prepared then measured by digital PCR. The analysis results revealed accurate copy numbers regardless of excess rcDNA in the reaction (R2=0.9913) (Fig. 1C). This finding indicated that the digital PCR method to quantify cccDNA is suitable for specific examination of cccDNA in clinical serum and liver specimens, in which rcDNA is in great excess. In addition, intrahepatic cccDNA was extracted from HepAD38 cells, where the whole HBV genome was integrated into the chromosome, and the virus genes were transcribed under control of the tet-off inducible promoter. The results of digital PCR analysis showed that the cccDNA copy numbers corresponded highly to the input amounts (R2=0.9931) (Fig. 2A). The cccDNA levels measured by digital PCR were highly correlated with those measured by real-time quantitative PCR (R2=0.9947), indicating that the digital PCR method was compatible with quantitative PCR but had higher sensitivity and was readily applicable to cccDNA analysis in clinical specimens (Fig. 2B).
 Various amounts of HBV cccDNA extracted from HepAD38 cells were quantified by digital PCR. The cccDNA copy numbers revealed by digital PCR were highly consistent with the input numbers (R2=0.9931). (B) Correlation of the cccDNA levels measured by digital PCR and real-time PCR. The results produced by the two methods were highly correlated (R2=0.9947).")
Detection of intracellular cccDNA in HepAD38 cells. (A) Various amounts of HBV cccDNA extracted from HepAD38 cells were quantified by digital PCR. The cccDNA copy numbers revealed by digital PCR were highly consistent with the input numbers (R2=0.9931). (B) Correlation of the cccDNA levels measured by digital PCR and real-time PCR. The results produced by the two methods were highly correlated (R2=0.9947).
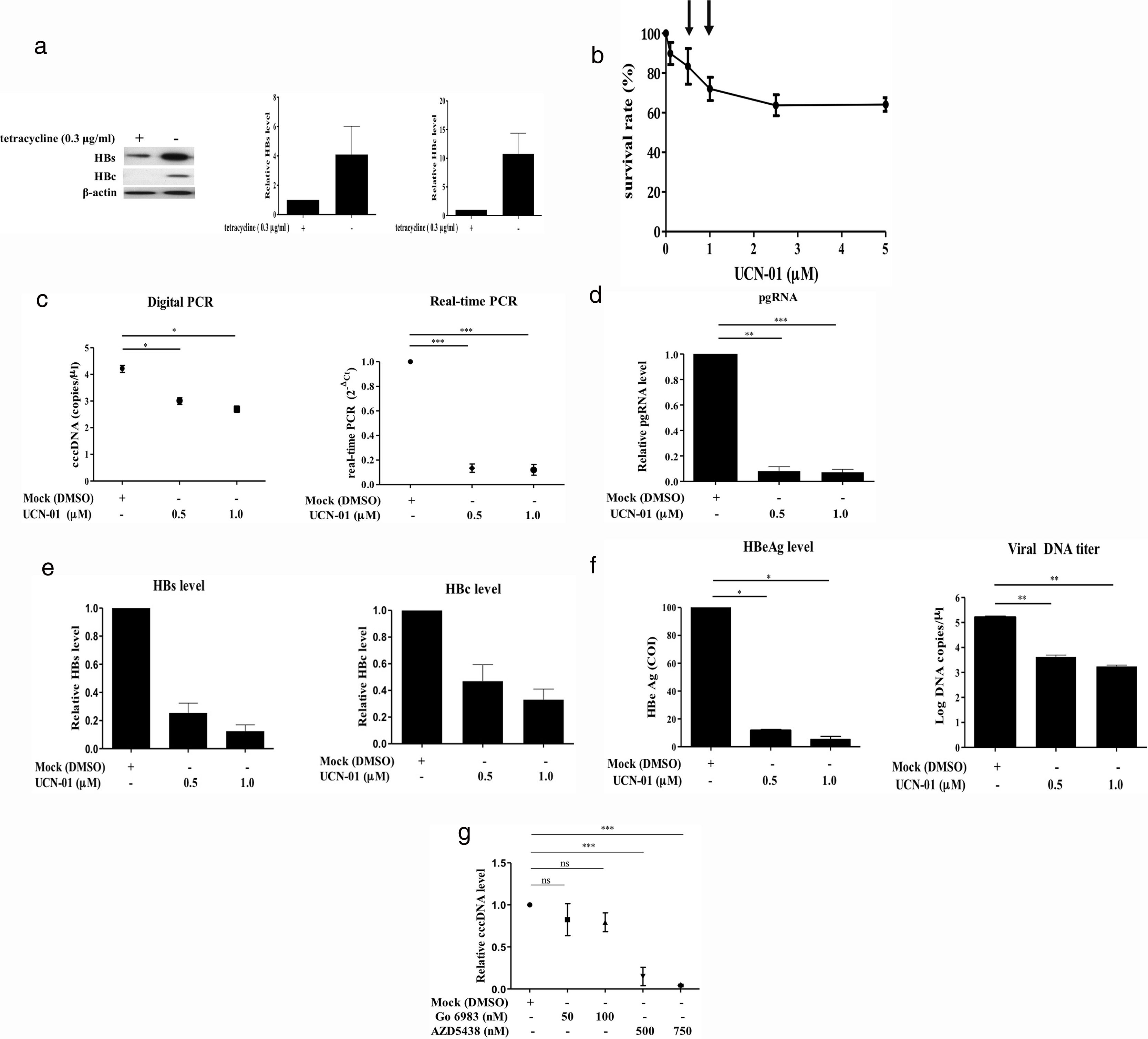
Recent studies have suggested that the host cell cycle machinery controls HBV replication [28]. Therefore, we examined whether HBV cccDNA synthesis is modulated by cell cycle progression. Virus-producing HepAD38 cells were first treated with UCN-01, an inhibitor with a broad-spectrum activity for PKC & CDK proteins, and then the intracellular cccDNA was purified and quantified by digital PCR (Fig. 3A). The results showed that treatments with UCN-01 greatly decreased cccDNA levels even in the concentrations (0.5 and 1μM) where the cell viabilities were not greatly compromised, as analyzed by MTT assays (Fig. 3B and C), excluding the possibility that UCN-01-induced cccDNA blockage is caused by global DNA degradation attributed to cell apoptosis. Consistently with cccDNA blockage, the intracellular viral pgRNA and HBV core and surface proteins and the HBeAg and viral DNA in virions secreted in the cell growth medium also decreased after treatment with UCN-01 (Fig. 3D–F).
 Virus protein production in HBV-producing HepAD38 cells. Viral core and surface proteins were greatly induced with removal of doxycycline from the growth medium. Left panel, representative image of Western blotting for HBV surface (HBs) and core (HBc) proteins. Right panel, summary of the results of at least 3 independent experiments. (B) MTT assays to detect the cell viabilities after UCN-01 treatments. UCN-01 concentrations of 0.5 and 1μM (arrows) were chosen for further studies of cccDNA. (C) Inhibition of the cccDNA levels after treatments with UCN-01, as measured by digital PCR and real-time PCR. (D–F) The intracellular HBV pgRNA (D) and the core and surface proteins (E) and the HBeAg and viral DNA levels in the growth medium (F) after UCN-01 treatments were analyzed. UCN-01 inhibited cccDNA synthesis as well as viral gene expression and virion production. (G) Intracellular cccDNA levels after treatments with the PKC inhibitor Go 6983 (50 and 100nM) and the CDK inhibitor AZD-5438 (500 and 750nM). AZD-5438 dramatically blocked cccDNA synthesis, whereas Go 6983 did not show a significant effect on cccDNA. *, **, and ***: p value<0.05, <0.01, and <0.001, respectively. ns: not significant.")
Inhibition of intracellular cccDNA synthesis by CDK inhibitors. (A) Virus protein production in HBV-producing HepAD38 cells. Viral core and surface proteins were greatly induced with removal of doxycycline from the growth medium. Left panel, representative image of Western blotting for HBV surface (HBs) and core (HBc) proteins. Right panel, summary of the results of at least 3 independent experiments. (B) MTT assays to detect the cell viabilities after UCN-01 treatments. UCN-01 concentrations of 0.5 and 1μM (arrows) were chosen for further studies of cccDNA. (C) Inhibition of the cccDNA levels after treatments with UCN-01, as measured by digital PCR and real-time PCR. (D–F) The intracellular HBV pgRNA (D) and the core and surface proteins (E) and the HBeAg and viral DNA levels in the growth medium (F) after UCN-01 treatments were analyzed. UCN-01 inhibited cccDNA synthesis as well as viral gene expression and virion production. (G) Intracellular cccDNA levels after treatments with the PKC inhibitor Go 6983 (50 and 100nM) and the CDK inhibitor AZD-5438 (500 and 750nM). AZD-5438 dramatically blocked cccDNA synthesis, whereas Go 6983 did not show a significant effect on cccDNA. *, **, and ***: p value<0.05, <0.01, and <0.001, respectively. ns: not significant.
The main groups of molecules whose kinase activities are inhibited by UCN-01 are PKC and CDK factors, suggesting that PKC and CDK are potentially important players in cccDNA synthesis [29]. To test this hypothesis, virus-producing HepAD38 cells were treated with (1) Go 6938, an inhibitor for a wide spectrum of PKCs, including PKCα, PKCβ, PKCγ, PKCδ, and PKCζ, that have been shown to involve in cell growth and differentiation [30]; and (2) AZD-5438, a potent inhibitor of CDK1, CDK2, and CDK9, which exhibited antiproliferative activity in human tumor cell lines [31]. The cccDNA levels in the cells treated with these drugs were measured by quantitative PCR. While the PKC inhibitor Go 6938 did not show a significant inhibitory effect on cccDNA synthesis, the CDK inhibitor AZD-5438 dramatically decreased cccDNA levels in a dose-dependent manner (Fig. 3G). These findings clearly demonstrated that CDK activity is required for HBV cccDNA synthesis.
4DiscussionChronic HBV infection is the major etiological factor of HCC. Viral replication activity has been documented to stimulate carcinogenic pathways in the liver [32]. As the main viral replication reservoir, HBV cccDNA plays a key role in HBV persistence [16]. Understanding the molecular mechanism of cccDNA synthesis is a very important task for paving the way to develop new-generation antivirals complementing NA drugs, which are highly effective but present with high rates of relapse after treatment discontinuation. The molecular mechanism of cccDNA synthesis remains obscure because reliable detection of cccDNA with high sensitivity and specificity has been challenging [20]. Here, by utilizing the new approach of digital PCR, we developed a quick and cost-effective method for precise quantification of cccDNA. With this technique, studies of the stepwise molecular mechanisms regulating cccDNA synthesis can be greatly improved in the near future.
HBV cccDNA is hypothesized to be an important predictive biomarker for the risk of adverse CHB outcomes. HBV cccDNA in the liver has also been shown to be highly correlated with serum ALT levels and liver inflammation, which ultimately leads to fibrosis, cirrhosis, and HCC [33]. cccDNA in hepatocytes also promotes the production of the viral oncoproteins HBx and LHBs, triggering oncogenic processes [34–37]. The traditional gold standard method for cccDNA detection is Southern blotting, which separates rcDNA and cccDNA by electrophoresis based on the different conformations of the two molecules and allows cccDNA visualization [38]. However, the experimental procedures for Southern blotting are time consuming and tedious and require a large amount of specimen, which make it impossible for clinical practice. Later, development of a gene amplification approach using cccDNA-specific PCR primers and probes combined with exonucleases specifically cleaving rcDNA, which contains some single-stranded regions, has greatly improved the sensitivity and specificity of cccDNA detection [39]. Furthermore, the recent revolutionary digital PCR technology provides absolute quantification of target nucleic acids without the need for an external calibrator and is therefore a robust approach for cccDNA detection [21].
Hepatitis B virus has been reported to dysregulate the cell cycle to promote viral replication and a premalignant phenotype [28]. In this study, we found that the broad spectrum PKC and CDK inhibitor UCN-01 dramatically decreased cccDNA synthesis. Further investigation also identified that CDK but not PKC was required for cccDNA synthesis. A previous study found that HBV-infected primary human hepatocytes (PHHs) were enriched in the G2/M phase, rather than G0/G1 phase, where cultured PHHs predominantly reside in, indicating that host cell cycle progression is likely interrupted by HBV infection [28]. cccDNA, the template of HBV transcription, plays a key role in the viral life cycle and permits infection persistence. Nuclear cccDNA accumulates in the hepatocyte nucleus as a stable minichromosome organized by histones and other chromosome-modulating cellular proteins [12]. These proteins are differentially expressed in various cell cycle phases, indicating that cell cycle machinery is functionally linked with cccDNA synthesis. Our data indicated that the CDK1, 2 and 9 inhibitor AZD-5438, an effective anticancer drug for solid malignancies, greatly diminished cccDNA synthesis in hepatocytes stably propagating virions. The stepwise molecular mechanism for how host cell cycle control participates in cccDNA synthesis remains to be deciphered; thus, the current finding has suggested that disrupting host cell cycle progression is a promising strategy to eradicate the HBV cccDNA pool in hepatocytes, leading to a virological cure.
In conclusion, this study developed a high-sensitivity and high-specificity method to precisely quantify HBV cccDNA in hepatocytes stably propagating viruses. The new digital PCR approach allows detection of cccDNA with a 1000-fold lower limit of detection than quantitative PCR. We also identified that host CDK activities are required for cccDNA synthesis. Future applications of digital PCR in clinical specimens in HBV carriers can be applied to drug screening for effective cccDNA inhibitors.AbbreviationsCCC covalently closed circular cyclin dependent kinase chronic hepatitis B digital PCR hepatocellular carcinoma limit of detection limit of quantitation limit of linearity (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tetrazolium nucleotide/nucleoside analogs sodium taurocholate cotransporting polypeptide polyethylene glycol primary human hepatocyte protein kinase C relaxed circular DNA reverse transcription-polymerase chain reaction SAM domain and HD domain-containing protein 1 tyrosyl-DNA-phosphodiesterases
CYB and CJH developed digital PCR and performed molecular and cellular experiments and drafted the manuscript; YWC and CYF performed digital PCR experiments; CJH performed statistical analyses of the experimental data; and WH designed the study. All authors read and approved the final manuscript.
FundingThis study was supported by the Taiwan Ministry of Science and Technology (grant nos. 107-2622-B-006-003-CC2, 106-2320-B-006-048-MY3 and 107-2320-B-006-032-MY3 to WH), the Taiwan Ministry of Health and Welfare (grant no. MOHW108-TDU-B-211-113003 to WH), and the National Cheng Kung University Center of Infectious Disease and Signaling Research (grant no. D105-22004 to WH).



