





P-713 - LEIOMIOSARCOMA RETROPERITONEAL EN PACIENTE CON SÍNDROME DE LI-FRAUMENI
Hospital Puerta de Hierro, Majadahonda.
Introducción: Los leiomiosarcomas retroperitoneales son tumores muy poco frecuentes que a veces se encuentran asociados a síndromes como Li-Fraumeni. El síndrome de Li-Fraumeni es una entidad de predisposición genética al cáncer, con herencia autosómica dominante.
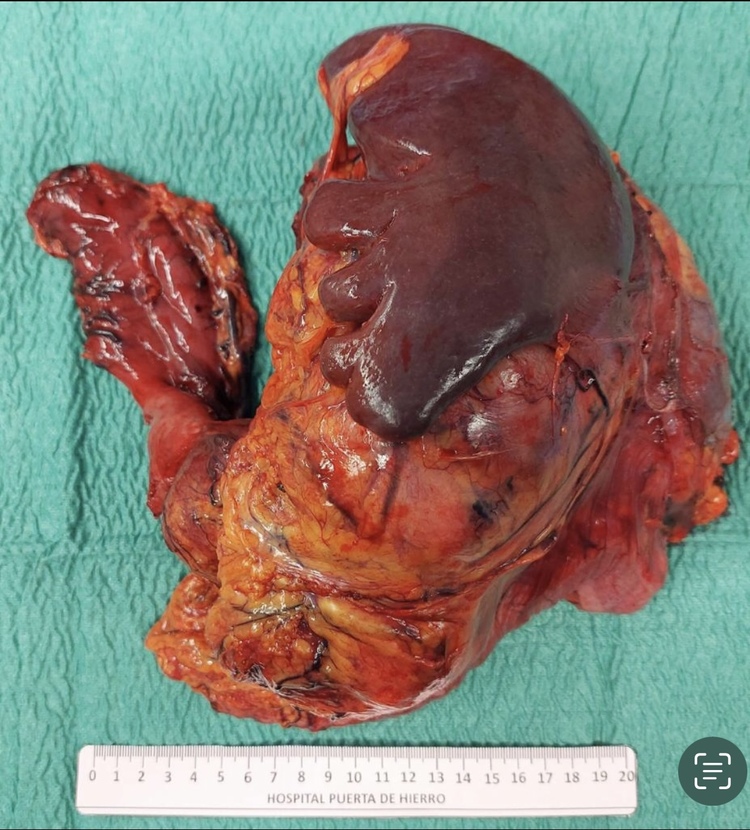
Caso clínico: Mujer de 38 años con los siguientes antecedentes: síndrome de Li-Fraumeni. Osteosarcoma de tobillo derecho: amputación infrapatelar. Carcinoma infiltrante ambas mamas: mastectomía radical bilateral. Acude por tumoración en flanco y fosa iliaca izquierda de tres semanas de evolución tras cesárea. A la exploración se palpa tumoración firme de 10 × 12 cm, móvil y no dolorosa. Tras este hallazgo se realizó como pruebas: 1) TAC toracoabdominal: gran masa sólida heterogénea de 15 × 9 cm que podría depender del cuerpo - cola del páncreas y que contacta ampliamente con el hilio esplénico, cara anterior del polo superior renal izquierdo y suprarrenal. 2) Biopsia: leiomiosarcoma. Tras estos hallazgos se decide cirugía: gastrectomía parcial, pancreatectomía corporocaudal, esplenectomía, nefrectomía y suprarrenalectomía izquierdas, hemicolectomía izquierda. Reconstrucción Billroth II y anastomosis transverso-sigma. El diagnóstico anatomopatológico: leiomiosarcoma retroperitoneal pT4, márgenes libres. El posoperatorio transcurrió sin complicaciones. Tras dos años de la cirugía, la paciente presenta metástasis hepáticas y se encuentra en tratamiento con quimioterapia.
Discusión: El síndrome de Li-Fraumeni es un síndrome hereditario que asocia una anormalidad en la proteína P53 originando la aparición de múltiples tumores primarios. En los portadores de la mutación se estima que el riesgo de desarrollar cáncer a los 30 años es del 50% y llega al 90% a los 60 años. En niños, el tumor maligno más frecuente es el osteosarcoma. En adultos, el cáncer de mama seguido de los sarcomas. Los sarcomas retroperitoneales forman un grupo de tumores muy poco frecuentes, donde el leiomiosarcoma ocupa el segundo lugar por detrás del liposarcoma. El único tratamiento potencialmente curativo es la extirpación en bloque del tumor con márgenes libres confirmados microscópicamente (resección R0). La resección completa solo es posible en el 20-50% de los casos ya que la mayoría son irresecables (por el gran tamaño, la complejidad, la invasión de órganos adyacentes...). Estas cirugías presentan una importante morbimortalidad, con tasas de complicaciones mayores en torno al 30% y mortalidad en la primera cirugía de aproximadamente el 5%. El uso de la radioterapia y la quimioterapia como tratamiento adyuvante o neoadyuvante todavía no está definido. El pronóstico del leiomiosarcoma es fatal, con una supervivencia a los cinco años del 0-29%, puesto que alcanza un gran tamaño y tiene una alta tasa de desarrollo de metástasis a 5 años de más del 50%.

