




To determine the effect of storage duration on cryopreserved ovarian tissue using fresh and frozen-thawed samples.
METHODS:Seventeen fertile patients underwent an ovarian biopsy during elective laparoscopic tubal ligation. The tissue sample was divided into three parts: one part was processed fresh (FG), and two were slowly frozen, cryopreserved for 30 (G30) or 180 days (G180), thawed and analyzed. Follicular density, follicular viability, and steroidogenic capacity were assessed.
RESULTS:We observed no differences between the groups in follicular density, which was assessed in hematoxylin and eosin–stained tissue sections. A heterogeneous follicular distribution was observed in the parenchyma, with a mean density of 361.3±255.4, 454.9±676.3, and 296.8±269.0 follicles/mm3 for FG, G30 and G180, respectively (p = 0.46). Follicular viability was greater in FG (93.4%) when compared with the cryopreserved tissues (70.8% for G30 (p<0.001) and 78.4% for G180 (p<0.001)), with no difference in viability between the frozen samples (p>0.05). The steroidogenic capacity of the tissue was not significantly reduced following cryopreservation.
CONCLUSION:The slow freezing procedures used for ovarian cryopreservation are capable of preserving follicular viability and maintaining the steroidogenic capacity of the tissue despite a roughly 30% decrease in follicular viability. Furthermore, short-term storage of ovarian tissue does not appear to compromise follicle integrity.
As a result of earlier diagnoses and appropriate treatments, an increasing number of patients are surviving cancer. However, the cytotoxicity of ionizing radiation and chemotherapeutic drugs frequently leads to premature ovarian failure, a condition with serious long-term consequences, including a reduced bone mass that can lead to osteoporosis, an increased incidence of cardiovascular disease, and the early occurrence of climacteric symptoms and infertility. The continuous evolution of assisted reproduction techniques has allowed us to develop various strategies to maintain reproductive function in female patients treated for neoplastic diseases, including hormonal manipulation and ovary transposition, as well as cryopreservation of embryos, gametes and ovarian cortical tissue.1
The primary objective of freezing of ovarian tissue is the preservation of ovarian function both in terms of fertility and hormone production, a goal that has not been attained using other methods. The freezing of ovarian tissue is currently used primarily for specific groups of patients for whom the remaining techniques are not recommended, such as the following: (1) prepubertal patients in which ovulation can not be induced for the conservation of embryos and gametes, (2) women without a partner who do not desire embryos obtained by fertilization with donor semen, (3) patients with estrogen-dependent neoplasms, such as breast cancer, and (4) women with malignant neoplasms requiring an immediate intervention for whom a delay in treatment initiation to induce ovulation might negatively impact the prognosis. In this last situation in particular, the collection of ovarian cortical tissue for cryopreservation is advantageous in that it can be performed at any time during the menstrual cycle and allows for the acquisition of hundreds of thousands of primordial follicles.2 Ernst et al.3 reported that one of their patients was transplanted with six fragments of ovarian tissue after experiencing a period of menopause and has since conceived naturally for a second time, giving birth to a healthy baby girl. This woman is the first reported patient to have had two children from separate pregnancies as the result of a transplant of frozen-thawed ovarian tissue. This result is encouraging and provides support for the development of better techniques to cryopreserve ovarian tissue to maintain the fertility of girls and young women undergoing gonadotoxic treatments. Considerable success has been achieved in terms of the restoration of patients' hormonal cyclicity, and to date, 15 healthy children have been born worldwide as a result of transplanted frozen-thawed ovarian tissue.4
When considering the reproductive possibilities of women treated for cancer, it is desirable to wait for a disease-free interval for a planned pregnancy. As a result, the ovarian tissue sample must often remain frozen for variable and possibly long periods of time. This is especially true for prepubertal female patients. Because the influence of the storage time on the magnitude of tissue damage has not been fully explored in cryopreserved ovarian tissue samples, the objective of the present study was to determine whether tissue damage progresses during sustained periods of cryopreservation by assessing the follicular viability and steroidogenic capacity of fresh ovarian tissue and that of frozen tissue tissues thawed after 30 and 180 days of cryopreservation.
MATERIALS AND METHODSPatients and study designThis was a prospective study conducted on 17 healthy patients undergoing elective tubal ligation via video laparoscopy. The mean patient age was 29.0±2.78 years, and all patients had at least two healthy living children. The study was approved by the local Research Ethics Committee, and all patients provided written informed consent to participate.
Ovarian tissue biopsies measuring approximately 2.0×2.0 cm were collected with laparoscopic scissors and prepared by immersion in nutrient M199 medium (Medium 199, Sigma; sterile filtered and supplemented with Earle's salts, L-glutamine and NaHCO3). For sample preparation, medullary tissue remnants, corpus luteum and eventual hemorrhagic cysts were eliminated by scraping with a surgical knife5 so that the tissue would have a maximum thickness of 2.0 mm with preservation of only the ovarian cortex for the appropriate penetration of the cryoprotectant solution. The tissue was then divided into three pieces and processed as follow: 1) fresh tissue analyzed directly (FG), 2) ramp frozen and thawed after 30 days (G30), and 3) ramp frozen and thawed after 180 days (G180).
Each fresh fragment was immediately processed and evaluated using hematoxylin and eosin (H&E.) staining to quantify the follicular density, LIVE/DEAD staining5 to measure the follicular viability, and tissue culture with measurement of steroids in the medium to quantify the steroidogenic capacity. Tissue culture was performed only in the last seven cases (Figure 1).
Histological evaluation
The first tissue fragment was fixed in 4% paraformaldehyde (Merck, UN 2213 Paraformaldehyd, 4.1, III) and embedded in paraffin. Serial, 4-μm-thick sections were cut and stained with &E. The stained sections were examined using light microscopy (400× magnification), and the number of follicles was counted. For the area calculations, a macro camera was used that permits the user to choose the previously captured image, generate its calibration (the pixel (image unit) × millimeter relationship), and determine the structural measurements.6 The total number of follicles per mm3 was calculated using the following formula: Nt = (No×St×t), where Nt is the number of follicles, No is the mean number of follicles observed in 1 mm2, St is the total number of sections in 1 mm3 of tissue, and t is the section thickness.7
ViabilityThe second tissue fragment was digested in an enzymatic solution for 45 minutes containing PBS (Dulbecco's PBS with Ca2+ and Mg2+ (1×), Invitrogen Gibco) and 1 mg/ml collagenase (Collagenase Crude Type IA Cell Culture∗T, Sigma) for analysis of follicular viability using a LIVE/DEAD Viability/Cytotoxicity Kit (Molecular Probes, Invitrogen). Although this level of structural evaluation was beyond the scope of the present study, we determined the mean follicular viability using a technique that indirectly assesses the functional capacity of the follicle. The LIVE/DEAD staining kit exploits the capacity of the cell to metabolize calcein using calcium as a co-factor. The produced metabolite fluoresces, and this fluorescence can be detected using microscopy. The cell must be viable and functional for this fluorescence to occur. The viability is reported as the percentage of viable follicles, with follicles considered viable when they contained a viable oocyte and more than 90% viable granulosa cells.8
In vitro cultureThe third tissue fragment was cultured in α-MEM (Minimum Essential Medium, Invitrogen) supplemented with 0.1% polyvinylpyrrolidone (PVP-40, Sigma), 100 ng/ml bovine insulin (Sigma), 10 ng/ml human recombinant IFG-1 (Invitrogen), 10-7 M androstenedione (Sigma), 11 mM non-essential amino acids (Gibco), 5 μg/ml human transferrin (Sigma), 1.4 ng/ml sodium selenite (Acros Organics), 10 mM sodium bicarbonate (Gibco), 0.02 M HEPES (Sigma), and antibiotics (10,000 IU penicillin and 10,000 IU streptomycin) (Sigma).9 Four fragments (2.0 mm×2.0 mm) from each patient were cultured per ml of medium in 4-well NUNC plates and incubated in a humidified 5% CO2 atmosphere at 37°C for 144 hours.10 Samples of the culture samples were taken at 48-hour intervals (0, 48, 96, and 144 hours of culture) and stored at -20°C until the progesterone (P4) and 17β-estradiol (E2) concentrations were determined using an ELISA (Siemens).
Cryopreservation of ovarian tissueThe tissue samples were equilibrated in three sequential freezing solutions containing medium 199 (Medium 199, Sigma; sterile filtered and supplemented with Earle's salts, L-glutamine and NaHCO3), 20% human albumin and 0.5 M (first step), 1 M (second step), or 1.5 M MS2SO4 (third step and freezing media), for a duration of 5 minutes each on ice. Following the last incubation, individual tissue samples were placed into cryovials containing 1 ml of freezing media. The tubes were moved to a controlled-rate freezer (Freeze Control System, CryoLogic, Victoria, Australia) with the temperature controlled by CryoGenesis V software (CryoLogic). The samples were cooled at 2°C/minute to -7°C, held for 10 minutes at -7°C, and when the actual temperature reached -7°C, manual seeding was performed using a cotton swab saturated with liquid nitrogen. Cooling was continued at 0.3°C/minute until the sample temperature reached -70°C. The tubes were then immersed and stored in liquid nitrogen (-196°C). The two frozen samples were thawed after 30 and 180 days and analyzed as described above. We used the rapid thawing method described by Gosden et al to thaw the samples.11
Statistical analysisAll data were analyzed using GraphPad 5.0 for Windows (GraphPad Software, San Diego, CA, USA). The mean and standard deviation of all variables were first calculated, and the normality was determined using the Kolmogorov-Smirnov test. The level of significance was set at p<0.05. The differences in the follicular density, follicular viability and estradiol and progesterone production in the culture medium at the different times studied (48, 96, and 144 hours for the identification of the peak production of these hormones) between groups were compared using the Kruskal-Wallis test followed by Dunn's multiple comparison test.
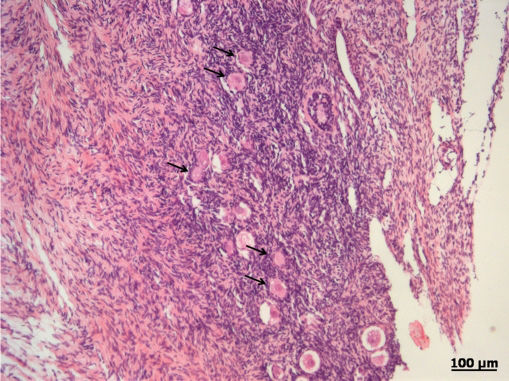
RESULTSA heterogeneous follicular distribution in the parenchyma, with a mean density of 361.3±255.4, 454.9±676.3, and 296.8±269.0 follicles/mm3 for FG, G30, and G180, respectively, was detected following H&E. staining (Figure 2). There were no differences observed between the groups (p = 0.46).

The follicular viability was higher in FG (93.4%) samples when compared with the cryopreserved samples (70.8% for G30 (p<0.001) and 78.4% for G180 (p<0.001)). There was no difference in the viability of the frozen samples (p>0.05).
The steroid levels in the culture medium at time zero were undetectable, confirming the absence of hormonal contamination during the manipulation of the medium. The average production of E2 and P4 during the seven days of culture did not decrease significantly after cryopreservation. There was no observable hormonal peak at the different culture times (48, 96, and 144 h) among samples of the same study group. Therefore, to more accurately represent the levels of these hormones, the mean estradiol (E2) and progesterone (P4) levels for each study group were calculated over all of the culture time points. The mean E2 levels were 2026±1782 pg/ml, 1272±1081 pg/ml and 849.6±366.2 pg/ml (p = 0.19) for FG, G30 and G180, respectively; and the mean P4 levels were 0.45±0.37 ng/ml, 0.26±0.08 ng/ml and 0.45±0.54 ng/ml (p = 0.86) for FG, G30, and G180, respectively.
DISCUSSIONIn this study, we aimed to determine the quality of frozen-thawed ovarian tissue after cryopreservation by assessing different aspects of the recovered tissue, including follicular density, follicular viability, and steroidogenic capacity. Follicular density refers to the number of follicles present in the tissue per unit volume and can vary both with patient age and within the same patient. Follicular density has been proposed by several authors as a quality control criterion to evaulate tissue after thawing.1,12–14 According to Poirot et al.2, a mean density of 30 to 400 follicles/mm3 tissue is expected in patients aged 20 to 30 years; however, it should be noted that follicle distribution is usually not homogeneous throughout the ovarian cortex. In the present study, we detected mean follicular densities ranging from 300 to 460 follicles/mm3, in agreement with the literature, and confirmed the heterogeneous distribution of the follicles. The objective of the assessment of follicular density was to guarantee that the various tissue samples would contain an adequate number of follicles, which would allow for the evaluation of functional status and viability, as the fresh and frozen-thawed tissue sections stained with H&E. were obtained from side-by-side fragments.
We also demonstrated the consistency of follicular density after tissue freezing and thawing, an expected result, as the loss of follicles due to the process would not have promoted a significant morphological change. To assess the real structural integrity of a follicle, it would have been necessary to examine markers of apoptosis or to perform an ultra-structural study by electron microscopy, procedures that were not performed as part of this study.
Previous studies examining the effects of cryopreservation on ovarian tissue have reported a follicular recovery rate and viability of 40% to 60% when frozen-thawed tissues were compared with fresh tissue.15,16 In the present study, slow ramp-freezing was used to preserve human ovarian biopsies, and this process preserved highly viable immature oocytes. Obviously, given the careful manipulation of the recently extracted tissue, the fresh samples were expected to demonstrate near 100% viability, a finding which was confirmed in our study.
While various degrees of damage to cryopreserved tissues have been presented by other authors,5,12,15 a recovery rate of approximately 60% viable follicles is considered acceptable5. Indeed, the 30% losses obtained in this study would be clinically significant compared with the possibility of using never-frozen tissues; however, in the clinical situations described (i.e., patients at risk for ovarian failure), the only options are cryopreservation of the tissue or gametes or no action at all. Obviously the potential gonadotoxicity of the chemotherapy to be used and the patients age must be taken into account. Ovarian tissue cryopreservation techniques should be offered only to patients at a high risk of ovarian failure; in all other cases, the less the ovary is manipulated, the better.
In this study, whatever damage occurred in the first 30 days appears unaltered after 180 days, as the percentage of viable follicles remained the same. We believe that the tissue damage occurs as a result of surgical manipulation and tissue processing, which may cause ischemia, and not the freezing process itself. Previous studies have shown that the majority of follicle loss occurs during the ischemic period before implantation and revascularization rather than during freezing and thawing. In sheep, it was estimated that approximately 7% of follicles are lost due to the cryopreservation procedure, whereas 60–70% are lost during the revascularization process in conjunction with transplantation.17 We found no data contesting or supporting our results in the literature, indicating that this is the first report to assess the influence of freezing time on tissue damage when using tissue cryopreservation techniques. Based on these findings, we can infer that after the sample is preserved, storage time should not decrease the quality of the tissue. Given that the clinical situations in which this technique is used usually require long periods of conservation, this information is extremely valuable for the management of these cases. Several authors have reported the implantation or use of samples preserved for different periods of time with promising results.18
In the same way that structural integrity does not reflect tissue viability, it also does not guarantee tissue functionality. We therefore assessed the steroidogenic capacity of the tissue, which in turn allowed us to infer the quality of its function in terms of hormone production. Ovarian tissue culture has been described by some authors as a way of assessing the functional capacity of ovarian tissue or of obtaining follicles in more advanced developmental stages for reproductive purposes.19,20 Sadeu et al.21 reported maintaining the viability of and inducing partial follicle maturation in human fetal ovarian cultures for up to 63 days. Thus, in light of the proven capacity for tissue survival in culture medium, we chose this model for functional evaluation of the cryopreserved tissue. It should be noted that one of the premises underlying the cryopreservation of ovarian tissue rather than isolated gametes is the possibility of maintaining the steroidogenic capacity of the tissue and not simply retaining the reproductive capacity.
In this study, the levels of estradiol and progesterone released by the ovarian tissue into the culture medium indicated that the steroidogenic capacity of the tissue was preserved. In addition, the cultured tissue continued to produce equivalent hormone quantities throughout the duration of culture and maintained the same rates of hormone production, indicating good tissue conservation during culture. It must be noted that the size of the cultured samples was standardized so that the tissue volume would not influence the hormone concentrations in the medium and thus generate a methodological bias.
In addition, hormone production did not differ between cultures of tissue frozen for 30 or 180 days, demonstrating that the capacity to produce hormones was preserved after cryopreservation, regardless of storage time. This result emphasizes the lack of further tissue damage due to the maintenance of low temperatures and supports the follicular viability findings, namely that the tissue damage is more likely due to the manipulation and processing of the sample than to the freezing process.
Finally, steroidogenic function does not necessarily indicate that reproductive function is maintained, as the ovary naturally first loses its reproductive capacity and then its steroidogenic capacity. This is best illustrated by normal ovarian physiology: fecundity abruptly decreases after 40 years of age (e.g., pregnancy rates of less than 5% after 40 years), whereas menopause occurs, on average, between 50 and 55 years of age.
Although ovarian tissue cryopreservation is still considered an experimental technique,1 many assisted reproduction centers have been routinely performing this procedure despite the lack of consensus statements concerning the selection of appropriate candidates22 or formal support from the relevant professional societies (e.g., the American Society of Oncology and the American Society for Reproductive Medicine) for its large-scale application.1 However, if the risk of ovarian failure related to oncologic treatment is high, then we believe that the patient should have autonomy in deciding whether or not to undergo the procedure after receiving in-depth information about the risks, benefits and effectiveness of the technique.
Many questions have arisen regarding ovarian tissue cryopreservation, such as the determination of the best candidates for this procedure, the best methods for tissue removal, the best form of freezing and thawing, whether one or both ovaries should be cryopreserved in full or in fragments, what the best site for re-implantation would be and whether vascular anastomosis should be performed. Further clinical and experimental studies are needed in order to address these concerns.
Despite the aforementioned points, we consider the technique to be adequate regarding the conservation of tissue and the maintenance of tissue viability and functional capacity, although the reproductive aspects of this procedure have yet to be explored. Nonetheless, there are enormous ethical implications involved in this type of evaluation. Only embryo fertilization, implantation and the birth of a healthy baby would confirm preservation of ovarian function and reproductive capacity.
Based on our findings, we conclude that although some loss in viability is observed after cryopreservation, the duration of cryopreservation does not interfere with the morphology or steroidogenic capacity of ovarian tissue. These findings permit the implementation of a safe and reliable standardized technique for human ovarian tissue freezing. It should be noted that the primary limitation regarding cryopreservation is currently the limited use of this tissue after thawing, with the published results still limited to case reports and studies examining small series of patients.
FUNDING:
This work was supported by the CNPq, Brazil.
No potential conflict of interest was reported.
Campos JR contributed to the development of freezing protocol and was also responsible for the sample cryopreservation (freezing-thawing), follicle isolation, DEAD-LIVE staining and manuscript preparation. Rosa-e-Silva JC was responsible for the sample collection (ovarian biopsy), statistical analysis and manuscript preparation. Carvalho BR contributed to the development of freezing protocol and was also responsible for the patient follow-up and manuscript preparation. Vireque AA was responsible for the sample cryopreservation (freezing-thawing), follicle isolation, DEAD-LIVE staining and manuscript preparation. Silva-de-Sá MF was responsible for the patient follow-up and manuscript preparation. Rosa-e-Silva ACJS contributed to the development of freezing protocol and was also responsible for the patient follow-up, sample cryopreservation (freezing-thawing), follicle isolation, DEAD-LIVE staining and manuscript preparation.