





Rubinstein-Taybi syndrome (RTS) is a rare developmental disorder characterized by craniofacial dysmorphisms, broad thumbs and toes as well as mental and statural deficiencies.1 RTS has a prevalence of 1 in 330,000 births2 and usually occurs sporadically, although it can be inherited as an autosomal dominant disorder.2,4 The diagnosis is based on characteristic features.3 The main clinical symptoms are failure to thrive, cardiac defects and recurrent respiratory infections.4 Other variable findings are colobomas, cataracts, renal anormalities3,4 and oro-dental anomalies.5 RTS patients also have an increased risk of developing neoplasias.6,7 Several tumors have been reported in RTS patients, including meningiomas, rhabdomyosarcomas, neuroblastomas, oligodendromas, seminomas, choristomas and leukemias.6,7
CREBBP and EP300 are the only genes associated with RTS.8,11 Microdeletions at band 16p13.3 occur in 10%–25% of RTS patients.9 Sequence analyses have detected CREBBP mutations in another 56% of affected individuals. Schorry et al. (2008)8 evaluated genotype-phenotype correlations in 93 RTS patients and performed complete sequencing of all 31 coding regions of the CREBBP gene. The authors concluded that there were no statistically significant differences in the classic dysmorphic findings of RTS patients (e.g., typical facial aspects and broad thumbs and toes) with and without CREBBP mutations. Mutations in EP300 have been identified in 3% of RTS patients.7,9
RTS typically occurs as a de novo mutation in a family.2 Most individuals represent simplex cases (i.e., the only affected member in a family); in most instances, the parents of an individual with RTS are not affected.2,4 In this case, the empiric recurrence risk for siblings is approximately 0.1%. Although individuals with RTS rarely reproduce, the theoretical risk for the offspring is 50%.2 Prenatal testing for at-risk pregnancies is possible if the disease-causing CREBBP mutation or deletion in the family is known.7
CASE DESCRIPTIONThe propositus, a 23-year-old Brazilian female patient, is the third child of healthy, non-consanguineous parents. The infant was born pre-term by Cesarean section. The birth weight was 1,900 g, the length was 47 cm, the occipitofrontal circumference (OFC) was 36 cm, and the body mass index was 31.35 (mild obesity). Developmental milestones were delayed: she sat without support at the age of 1, walked at 2.5 years and acquired control of the bowels at age 4. She spoke only a few meaningful words at the age of eighteen. The clinical history of the infancy and childhood was noteworthy, with feeding problems and recurrent respiratory infections (sinusitis, otitis medias, tonsillitis and pneumonias) from two months of age until the age of five. Splenomegaly was observed at 21 years of age, and a detailed immunological analysis of this patient was performed by Torres (2008)13. Persistent mild to moderate leukocytosis (13,000 to 26,000/mm3) was observed despite the absence of clinical signs of infection. Forty to fifty percent of blood neutrophils presented with scarce or absent granules. A myelogram also showed dysgranulopoietic granulocytes.
A physical examination at 23 years of age showed a weight of 61 kg (75Figure 1). She also had broad, radially deviated thumbs and broad halluces (Figure 2).


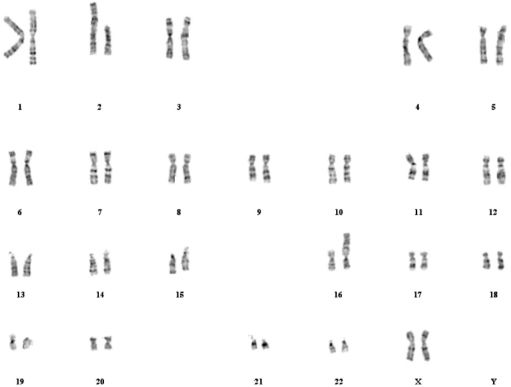
Chromosome preparations were obtained from peripheral blood lymphocytes from the propositus and her parents. Karyotyping with GTG banding was performed at the 450–550 band level using standard methods9 by the Fleury Laboratory. The patient’s karyotype showed a balanced reciprocal translocation between chromosomes 2 and 16 without visible deletion; the full karyotype is 46, XX, t(2;16) (q36.3; p13.3) (Figure 3). The parent’s karyotypes were normal, so this translocation was de novo. The recurrence risk for the parents is very low (approximately 0.1%).
DISCUSSION
Chromosomal rearrangements involving band 16p13.3 form the minority (0.6%) of CREBBP mutations.18 There have been seven reported cases of chromosomal rearrangements in RTS patients, and four of these were translocations.14–18
The 4-year-old RTS boy described by Petrij et al. (1995)17 presented with the same translocation (2; 16) with the same breakpoints as our patient. The common clinical findings of this boy and our patient were feeding problems, short stature and recurrent respiratory infections. To the best of our knowledge, our RTS patient is the third reported case with a de novo reciprocal t (2;16) (q36.3; p13.3).
We found no other studies describing RTS patients with dysgranulopoietic neutrophils. As RTS patients have an increased risk of hematological malignancies,6 appropriate laboratory testing and hematological follow-up are warranted.