




Introduction
Treatment of systemic infections caused by Salmonella and eradication of carrier state is not easy when the strain is multiresistant. The two main therapeutic alternatives against these organisms are both fluoroquinolones and new β-lactams, for their bactericidal activity and their excellent pharmacokinetic properties1-3.
The main mechanism of resistance to fluoroquinolones in gram-negative bacteria is caused by mutations in genes coding for DNA-gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE)4. Other mechanisms involved in resistance to these antimicrobials include altered expression of porins or lipopolysaccharide leading to decreased penetration of fluoroquinolones within bacteria, expression of efflux pumps for eliminating the compound from the cell5-7, and presence of the plasmid-mediated Qnr protein, which protects topoisomerase from the action of quinolones8,9. In Salmonella, mutations in genes coding for DNA-gyrase1-3,10-13, and more recently for parC14-16, have been described. The importance of hyperexpression of efflux pumps has also been proved3,17-19. Although altered outer membrane proteins have also been detected, a direct relationship between loss of porins and fluoroquinolone resistance in Salmonella has yet to be determined3.
Resistance to β-lactams in gram-negative bacteria is mainly due to β-lactamase expression4,20. Altered permeability, the presence of efflux pumps and altered penicillin binding proteins (PBP) also contribute to resistance. Ampicillin resistance in Salmonella enterica is often due to production of TEM-1, TEM-2, PSE-1, OXA-1 or SHV-1 β-lactamases. Extended-spectrum β-lactamases or plasmid mediated cephamycinases have uncommonly been reported23-25. Salmonella strains resistant to β-lactams19,23,24 or to quinolones1,3,13,26-28 have already been described, but strains simultaneously resistant to both classes of antimicrobial agents are infrequent. Development of resistance to these agents during therapy is also unusual17,28-30. Treatment failures with fluoroquinolones for infections caused by nalidixic acid-resistant and low-level fluoroquinolone resistant Salmonella strains have been documented31.
The objective of this study was to determine the mechanisms of resistance to fluoroquinolones and to β-lactams in strains of S. enterica isolated from two patients treated with fluoroquinolones.
Methods
Patient A
A 44-year-old male infected with the human immunodeficiency virus was treated with amoxicillin for an enteritis by S. enterica in August 1993 (strain not available). The patient developed a carrier state and received continuous norfloxacin therapy (500mg / 12h) for three years. From January 1994 to April 1996, the patient presented several episodes of gastroenteritis, and S. enterica was isolated each time (strains A1 to A10). A mutant (A10M) with a high level of fluoroquinolone resistance was selected from the last isolate (A10) using the Szybalski gradient method32.
Patient B
A 68-year-old male diagnosed of ulcerative colitis 30 years ago. Cholecystectomy was performed due to cholecystitis in 1985. In 1988, he was diagnosed of sclerosing cholangitis in the initial phase, and prophylactic norfloxacin (500mg / 48h) was administered. In August 1998, the patient was admitted to our centre for gastroenteritis by S. enterica (strain not available). He was treated with ciprofloxacin (500mg / 12h) for one week. In September 1998, the patient was readmitted for a septic infection of probable biliary origin, and S. enterica was cultured from both blood (strain not available) and stools (strain B1). Therapy with i.v. ciprofloxacin (400mg / 8h) was started and clinical evolution was favourable. The patient was discharged on norfloxacin (400mg / 48h) administered as selective intestinal decontamination for prevention of further cholangitis episodes. Between October and December 1998, he was admitted to the hospital on several occasions for a febrile syndrome of probable biliary origin. During this period, prophylaxis with ciprofloxacin (500mg / 24h) was administered. In December 1998, the patient was admitted to hospital for diarrhoea and fever, and S. enterica was cultured from few stool cultures (strains B2 and B3). Clostridium difficile toxin was also detected. This episode was treated with amoxicillin-calvulanic acid and metronidazole, and prophylaxis with ciprofloxacin was continued.
Serotyping
Determination of serotype of the studied strains was performed at the Centro Nacional de Microbiología, Virología e Inmunología Sanitarias (Majadahonda, Madrid, Spain).
Pulsed-Field Gel electrophoresis
DNA was extracted according to Rasheed et al33, and cut using the restriction enzyme XbaI (Roche, Mannheim, Germany). Samples run for 21h at 250V from 5 to 50 s, at 14 °C in 1% agarose gel containing 0,5x TBE (Trizma 0,1M, boric acid 0,1M and EDTA 0,2 mM) (Sigma, Madrid, Spain).
Susceptibility testing
Initial determination of susceptibility to antimicrobial agents was performed by disk-diffusion, following the NCCLS guidelines34,35. The minimal inhibitory concentrations (MICs) were performed by Etest (Biodisk, Solna, Sweden), according to manufacturer's recommendations. The following antimicrobial agents were evaluated: nalidixic acid, ciprofloxacin, clinafloxacin, levofloxacin, norfloxacin, ampicillin, cefotaxime, cefoxitin, ceftazidime, cefuroxime and tetracycline. MIC of chloramphenicol was determined by microdilution (Sensititre, Trek Diagnostic System, West Sussex, England)35.
The activity of the efflux pump inhibitor phenyl-arginyl-naphtylamide (PAN) (Sigma, Madrid, Spain) was evaluated. MICs of nalidixic acid (Sigma), pefloxacin (Rhône-Poulenc, St. Antoine, France), ciprofloxacin (Sigma) and norfloxacin (Sigma) with and without 20 mg/l of PAN against isolates A1, A3, A4, A6, A10, A10M, B1, B2 and B3 were determined by microdilution method, according to NCCLS guidelines35.
β-lactamases activity
Extracts containing β-lactamases were obtained as previously described36. β-lactamases were detected by colorimetry, by mixing 100 µl of crude β-lactamase extract and 50 µl of a solution containing 1mg/ml of nitrocefin (Oxoid, Hampshire, England).
Sequencing of the gyrA, gyrB, parC y parE genes
Primers and conditions described by Giraud et al2 were used for amplifying the quinolone-resistance determining region (QRDR) of genes gyrA, gyrB, parC y parE. Sequencing of the amplified products was performed using a fluorescent probe (Cy5 dideoxi) and an ALF Express sequencer (Automatic Laser Fluorescent DNA Sequencer, Amersham Biosciences, Uppsala, Sweden). Nucleotide and deduced aminoacid sequences were analysed with software available at the National Center for Biotechnology Information Internet site (www.ncbi.nlm.nih.gov).
Complementation assays
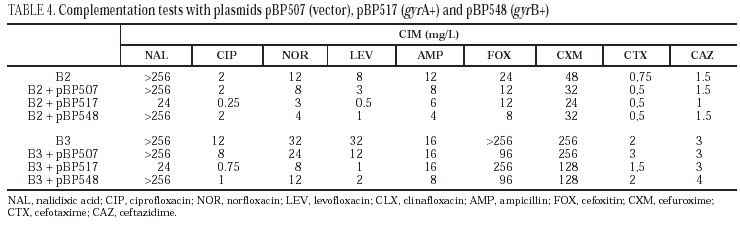
Plasmids pBP507 (vector), pBP517 (gyrA+) and pBP548 (gyrB+) were introduced in S. enterica B2 and B3 strains by transformation. These plasmids code wild-type gyrA and gyrB genes, allowing to restore susceptibility to quinolones in transformants when resistance in the parental strain is caused by mutations in gyrA or gyrB, respectively1,37.
Outer membrane protein (OMP) profiles
OMP were obtained from an exponential phase culture in 40 mL of Mueller-Hinton broth, using a previously described method38. Bacteria were separated by centrifugation (5.000 x g, 4 °C, 15 min) and were subsequently sonicated. Cell envelopes were recovered by centrifugation (13.000 x g, 4 °C, 45 min) and treated with lauryl-sarcosynate (Sigma). OMPs were obtained by means of an additional centrifugation step (13.000 x g, 4 °C, 45 min). OMP profiles were determined by electrophoresis in polyacrilamide-sodium dodecyl sulphate gel, and stained with coomassie blue. Salmonella choleraesuis subsp. choleraesuis NCTC 5188 was used as a control.
Determination of norfloxacin accumulation
Basal accumulation of norfloxacin was performed as previously described39. Bacteria were separated by centrifugation and resuspended in phosphate buffer at an optical density (520 nm) of 1,5. The bacterial suspension was incubated with norfloxacin (10 mg/l) at 37°C for 30 min. Bacteria were separated from extracellular fluid by differential centrifugation through a silicon oil layer (density, 1,029 g/cm3) and lysed in 0,1 M glycine-HCl buffer (pH 3,0). Samples were centrifuged at 12.800 x g for 5 min and the accumulation of norfloxacin was measured by spectrofluorometry (Hitachi 2000, Tokyo, Japan). In another series of parallel experiments, the effect of carbonyl-cianide-m-chlorophenilhydrazone (CCCP, 0,1 mM, Sigma) was tested. For this purpose, after bacteria were pre-incubated with norfloxacin (10 mg/l) for 10 min, the metabolic inhibitor was added and an additional incubation of 20 min was performed. Experiments were done in duplicate, three times, on different days.
Results
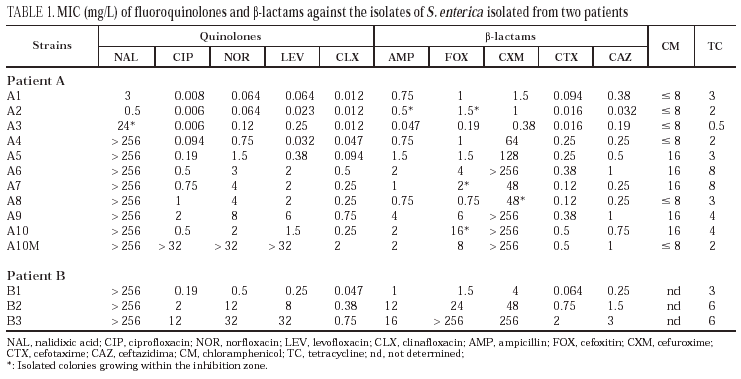
All isolates from patient A were identified as S. enterica subspecies I. They were non-mobile and had a rough morphology, which precluded serotyping. Isolates from patient B corresponded to serotype Enteritidis. Identical restriction patterns were obtained by PFGE between isolates from the same patient and between both patients. There was no clinically apparent relationship between the two patients. Results of susceptibility to the evaluated antimicrobial agents are presented in table 1. Isolates from patient A were divided into four groups: the first group was constituted of isolates A1 and A2 (isolated in 1994) which were susceptible to all antimicrobial agents. The second group included the A3 isolate. It was isolated in February 1995 and showed a moderate resistance to nalidixic acid, a moderate decrease in susceptibility to norfloxacin and levofloxacin and susceptibility to ciprofloxacin and clinafloxacin. A small increase in susceptibility to β-lactams was also observed. Isolates A4 and A5, obtained early in 1995, are included in the third group; they are resistant to nalidixic acid and the MICs of fluoroquinolones against them are higher; they also showed resistance to cefuroxime and presented a moderate increase in the corresponding MICs of both cefotaxime and ceftazidime. Finally, the fourth group included isolates A6 to A10, cultured towards the end of 1995 and the beginning of 1996. In the latter two isolates, the increased resistance to fluoroquinolones was more evident, and variations in the activities of cefuroxime and other β-lactams were also noted.
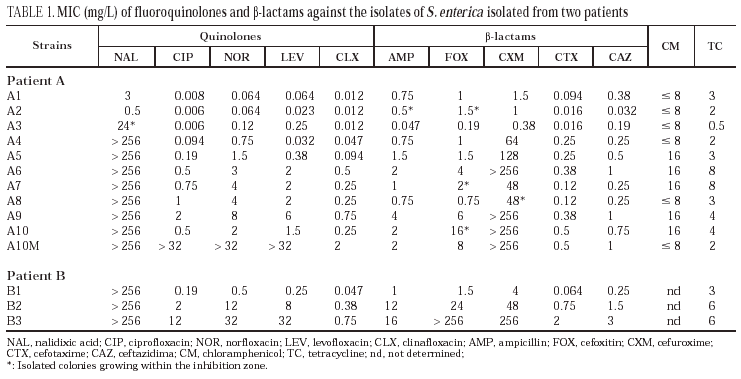
All isolates from patient B (B1-B3) were resistant to nalidixic acid. The MICs of both fluoroquinolones and β-lactams against isolates B2 and B3 were elevated.
In order to evaluate the possible mechanisms involved in resistance to β-lactams and fluoroquinolones the following approach was used:
1. The presence of a β-lactamase, which would explain β-lactam resistance, was ruled out.
2. Mutations in the QRDR of the DNA-gyrase and topoisomerase IV encoding genes that would account for resistance to fluoroquinolones were investigated.
3. The presence of altered outer membrane proteins and/or the expression of an active efflux mechanism that would be related to resistance to both classes antimicrobial agents were disregarded. Results from these studies are shown in Tables 2 and 3.
None of the isolates from the two patients evaluated presented β-lactamase activity.
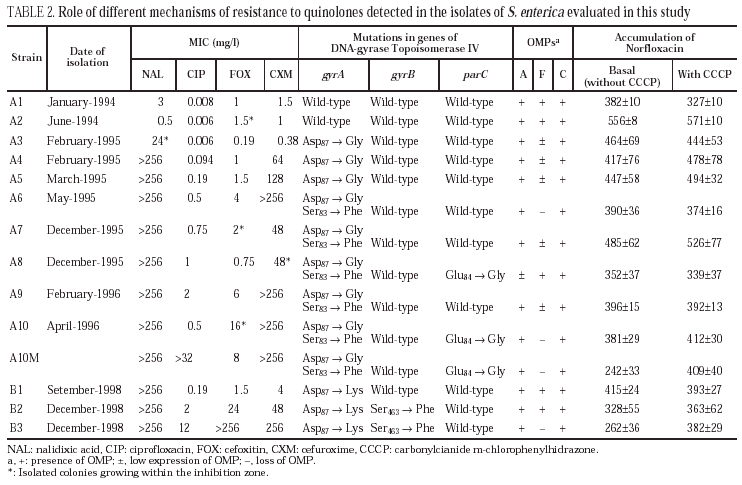
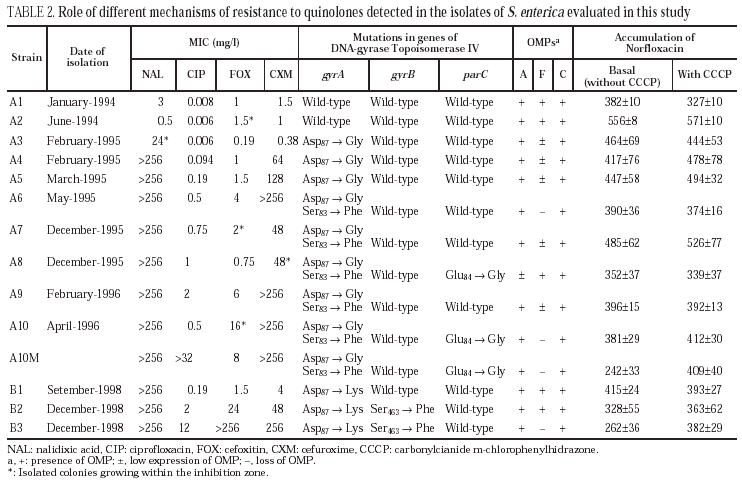
Isolates A3, A4 and A5 presented a mutation in gyrA (Asp87 → Gly). In isolates A6, A7 and A9, a second mutation in gyrA (Ser83→Phe) was also observed. Finally, isolates A8 and A10 (and A10M) showed a third mutation in gene parC (Glu84 → Gly).
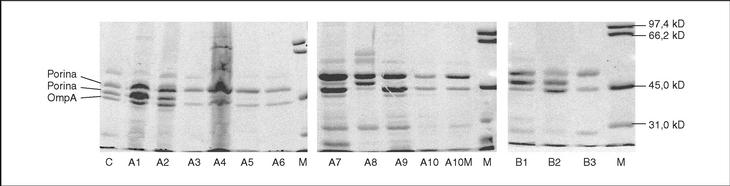
All isolates presented two or three OMP (fig. 1). As a result of electrophoretic motilities and in comparison with previous descriptions in Salmonella and other enterobacteria, all isolates presented a band corresponding to OmpA (the protein of lower molecular weight), and one or two porins. Expression of OmpA was reduced in isolate A8. Isolates A1, A2 and A8 showed two porins; a decreased expression of the low molecular weight porin (corresponding to OmpF) was noted in isolates A3, A4, A5, A7 and A9, and a complete loss of this porin occurred in isolates A6, A10 and A10M (fig. 1).
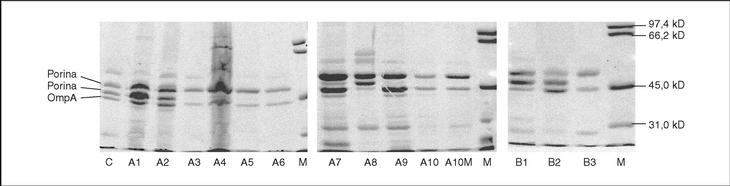
Figure 1. SDS-PAGE of outer membrane proteins of different S. enterica isolated from patients A and B.
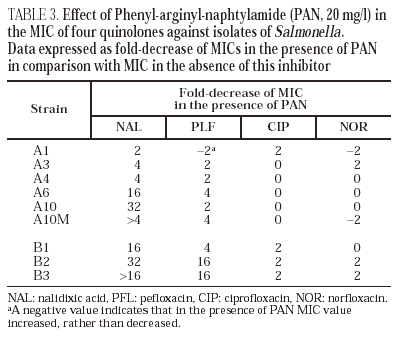
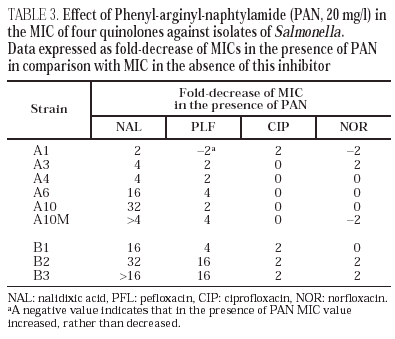
Norfloxacin accumulation showed variability in basal values, ranging from 328 ± 10 to 556 ± 8 ng/mg of dry bacterial weight, the highest value corresponding to isolate A2 (table 2). In the presence of CCCP, norfloxacin accumulation showed a significant increase in comparison with basal levels in isolate A10M. Results from this experiment suggest an active efflux mechanism in this isolate. In the presence of PAN, MICs of norfloxacin did not change (three out of the six evaluated isolates), or even increased twofold (two of the six isolates). Similarly, MICs of ciprofloxacin did not change (five of the six isolates) or decreased only twice (the remaining isolate). On the contrary, PAN caused a significant decrease (of at least four times) of the MICs of nalidixic acid (five of the six isolates) and of the MICs of pefloxacin (two of the six isolates) (table 3).
All isolates from patient B showed a mutation in gyrA (Asp87 → Lys). In B2 and B3 a second mutation in gyrB (Ser463 →Phe) was also noted. Loss of OmpF and a significant increase in the accumulation of norfloxacin in the presence of CCCP in comparison with the basal level was only observed for isolate B3 (fig. 1) (Table 2). PAN decreased the MICs of nalidixic acid (16 to 32 times) and pefloxacin (4 to 16 times), but in the presence of this inhibitor, MICs of norfloxacin or ciprofloxacin did not change or were only twice reduced (Table 3).
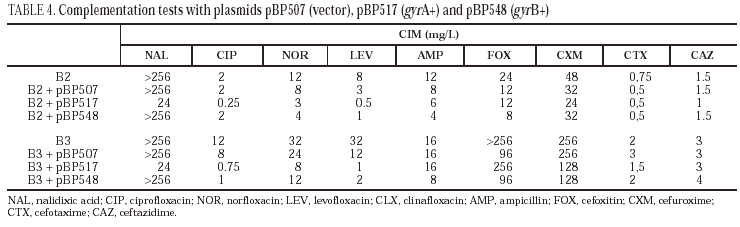
Complementation studies demonstrated that the introduction of plasmid pBP517 (coding for wild-type gyrA) in isolates B2 and B3 decreased the MIC of nalidixic acid, ciprofloxacin, norfloxacin and levofloxacin (Table 4). Similarly, transformation of isolates B2 and B3 with plasmid pBP548 (coding for wild-type gyrB) moderately reduced the MIC of norfloxacin and of levofloxacin in isolate B2, and the MIC of ciprofloxacin and levofloxacin in B3. MICs of the three fluoroquinolones evaluated were two to three times higher against the transformants from isolate B3 than against transformants from isolate B2. Expression of wild-type GyrA or GyrB from the corresponding plasmids did not significantly affect (as could be predicted) the MICs of the tested β-lactams.
No mutations in parE in any of the isolates from either patient were found.
Discussion
Salmonella infections continue to be a challenge in Spain25,40,41 and it is thus important to study the resistance mechanisms to antimicrobial agents in species of this genus and their clinical impact.
Both patients in this study received long-term treatment with fluoroquinolones and Salmonella isolates with increasing resistance to fluoroquinolones and β-lactams were obtained. Phenotypic traits, along with genotypic information (mutations detected), support the clonal relationship of the isolates obtained in both patients. Previous studies have demonstrated that changes in topoisomerases are insufficient to explain the level of resistance to quinolones in S. enterica13. In agreement with this observation, our results showed that multiple resistance mechanisms were involved in the strains we have evaluated, and included changes in topoisomerases (mutations in genes gyrA and perhaps parC, in patient A; and in genes gyrA and gyrB in patient B), altered permeability (mainly because of reduced expression of porin OmpF, in both patients), and expression of an active efflux. The most frequently described mutations in S. enterica11 have been Asp87 → Gly and Ser83 →Phe in gyrA. Mutation Asp87 → Gly apparently confers resistance only to nalidixic acid. In Escherichia coli, high-level resistance to fluoroquinolones requires a second mutation in the same gene and/or a mutation in parC37. In our case, the MIC of ciprofloxacin against isolates with two mutations in gyrA increased 2-4 times or 8-12 times in comparison with isolates A5 or A3, respectively, both with a single gyrA mutation. It does not seem plausible that the mutation Glu84 →Gly in parC observed in clinical isolates A8 and A10 significantly contributes to resistance, as in both isolates no increase in the resistance in comparison with isolates A7 or A9 (lacking this mutation) was observed. In patient B, in addition to mutation in gyrA (Asp87 →Lys), a second mutation in gyrB (Ser463 →Phe) was also noted. The aminoacid change Ser → Tyr because of a mutation in the same codon has been described by Gensberg et al10 in S. typhimurium. The complementation test demonstrated the importance of the mutation in gyrA in the resistance of isolates from patient B to both nalidixic acid and fluoroquinolones. Using the same approach, we observed that the mutation in gyrB was also related to quinolone resistance. These findings agree with those of Heisig1 who demonstrated that high level resistance of Salmonella to quinolones could be due to mutations in either gyrA or gyrB. Results on the effect of porin expression in the level of resistance are not conclusive. When we compared antimicrobial agent activities against two pairs of isolates differing in the level of OmpF expression (A6 and A7 on one side and A9 and A10 on the other side). We observed that isolates deficient in this porin were slightly more resistant to β-lactams, but not to quinolones. In any case, decreased expression or loss of OmpF does not completely explain the increased resistance to cefuroxime, cefoxitin and cefotaxime observed in some isolates. Considering that these isolates lack β-lactamase, they perhaps contained additional mechanisms of resistance to β-lactams (altered PBPs?). In both Klebsiella pneumoniae38,42 and E. coli (unpublished data), loss of only one of the major porins is of little clinical relevance in resistance to β-lactams and quinolones, presumably because the expression of the other porin allows sufficient penetration of antimicrobial agents to inhibit the microorganism. Further studies using genetic methods to evaluate the re-expression of the lost porin are needed to know the real impact of this mechanism on resistance, and to evaluate the relevance of these channels in the penetration of different groups of antimicrobial agents.
The fact that accumulation of norfloxacin in isolates A10M (in vitro derived mutant) and B3 (selected in vivo) increased when the pump mechanism was inhibited by CCCP suggests that both isolates showed an increased activity of active efflux. It is interesting to note that although the expression of an efflux system is related with an increase in the level of resistance to quinolones, it does not affect other families of antimicrobial agents, such as β-lactams, tetracycline or chloramphenicol.
The transformants from B2 were more susceptible than those from B3, when either wild type gyrA or wild type gyrB were transformed. This suggests that the loss of one porin and expression of an active efflux mechanism in B3 caused decreased accumulation of quinolones, and consequently a decrease in their activity. In E. coli6,17 and in S. typhimurium43, the main efflux system is AcrAB, regulated by multiple mechanisms. Operons marRAB and soxRS that regulate acrAB, were also implicated in the control of the expression of porin OmpF. Several mutations in this regulatory system causing increased AcrAB expression and decreased OmpF production have been described43. This is similar to the situation observed in isolate B3 from patient B (fig. 1). The low impact of this double mechanism on MIC has already been described by Piddock et al27. Expression of other porins, through which antimicrobial agents may still penetrate in the absence of OmpF would explain this situation, as recently demonstrated in K. pneumoniae strains44.
MICs of nalidixic acid decreased in the presence of PAN for five of the six isolates of patient A and for all three isolates of patient B, which also suggests that these isolates expressed an active efflux mechanism. A similar yet less evident effect was observed with pefloxacin, but the MICs of both ciprofloxacin and norfloxacin did not change, or changed minimally. The fact that the decreases induced by PAN were higher for nalidixic acid and pefloxacin, (two of the more hydrophobic quinolones) than for ciprofloxacin and norfloxacin, is compatible with the currently accepted theory that efflux pumps preferably recognise hydrophobic substrates in a poorly specific way. There are few data on the interaction of CCCP and PAN with active efflux pumps. It is also unknown whether both inhibitors are active against the same efflux system(s). The differences in the effects observed with PAN and with CCCP are possibly caused by the distinct ability of the two compounds to inhibit different active efflux systems that could be differently involved in the elimination of certain quinolones. It is also possible that the decrease in the accumulation of norfloxacin in the presence of CCCP has only been translated in a small increase in the MIC in strains carrying altered quinolone targets (as previously demonstrated in K. pneumoniae strains42). This could explain the very slight decrease of the norfloxacin MIC in the presence of PAN. These results were similar to other findings in our laboratory with K. pneumoniae strains for which the mechanisms of resistance to quinolones have been characterised (unpublished data). Further studies are needed to understand the active efflux mechanisms in enterobacteria and the true significance of the methodological approaches used to demonstrate this mechanism. Basal accumulation of norfloxacin in clinical isolates from patient A (where an active efflux mechanism was not observed) showed marked variations. It is interesting to point out that in isolate A2, where the basal accumulation of norfloxacin was highest, MICs of nalidixic acid and several quinolones were the lowest in the whole series. This may be linked to the method we used (presenting some degree of variation as indicated from standard deviation values obtained), but it can not be discarded that the precise tuning of active efflux in Salmonella could cause it. Additional studies are needed to clarify this aspect and to evaluate the real impact of efflux pumps in quinolone resistance in this species.

