



El síndrome de Ohtahara (SO, OMIM#308350, ORPHA1934) es una encefalopatía epiléptica de inicio precoz (EEIP) caracterizada por espasmos, crisis epilépticas intratables, un trazado electroencefalográfico de brote-supresión y retraso psicomotor grave. En la mayoría de los pacientes con SO se han identificado mutaciones en el gen STXBP1, que codifica para la proteína de unión a sintaxina 1 y que está implicado en el mecanismo de exocitosis de las vesículas sinápticas.
Paciente y resultadosSe presenta el caso clínico de un varón de 19 meses de edad diagnosticado de SO en el que se ha identificado una mutación no descrita (c.1249+2T>C, G417AfsX7) en el gen STXBP1. La mutación está localizada en uno de los sitios donadores implicados en el procesamiento del ARNm del gen, lo que produce la pérdida del exón 14 y el posterior truncamiento de la proteína que codifica.
ConclusionesEsta nueva mutación en el gen STXBP1, identificada en un paciente sin lesión cerebral estructural subyacente, amplía el espectro mutacional asociado a este devastador síndrome epiléptico.
Ohtahara syndrome (OS, OMIM#308350, ORPHA1934) is an early-onset epileptic encephalopathy (EOEE) characterised by spasms, intractable seizures, suppression-burst pattern on the electroencephalogram, and severe psychomotor retardation. Mutations in STXBP1 —a gene that codes for syntaxin binding protein 1 and is involved in synaptic vesicle exocytosis— has been identified in most patients with OS.
Patient and resultsWe report the case of a 19-month-old child with OS who displays a previously unreported mutation in STXBP1 (c.1249+2T>C, G417AfsX7). This mutation is located in a donor splice site and eliminates exon 14, resulting in a truncated protein.
ConclusionThis previously unreported STXBP1 mutation in a subject with Ohtahara syndrome and non-lesional magnetic resonance imaging (MRI) broadens the mutational spectrum associated with this devastating epileptic syndrome.
El síndrome de Ohtahara (SO, OMIM#308350, ORPHA1934) es una encefalopatía epiléptica de inicio precoz (EEIP) caracterizada por espasmos, crisis epilépticas intratables, un trazado electroencefalográfico de brote-supresión y grave retraso psicomotor1. Cuando los pacientes no presentan alteraciones cerebrales a nivel estructural ni existe un trastorno metabólico subyacente, se debe considerar la posibilidad de una etiología genética. En la mayoría de los pacientes con SO se han identificado mutaciones en el gen STXBP12,3.
El gen STXBP1 codifica para una proteína de unión a sintaxina 1 que regula la secreción de las vesículas sinápticas a través de la unión a las proteínas soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)4. Este complejo de proteínas tiene un papel esencial en la fusión de las vesículas sinápticas con la membrana plasmática presináptica, lo que permite la liberación de los neurotransmisores en la hendidura sináptica5.
En este trabajo se describe una nueva mutación en uno de los sitios donadores implicados en el procesamiento del ARNm del gen STXBP1 en un paciente con SO sin lesión cerebral subyacente.
Paciente y métodosEl paciente fue diagnosticado en la Unidad de Neuropediatría del Hospital Virgen de la Salud de Toledo y se derivó para su estudio genético al Laboratorio de Neurología del IIS-Fundación Jiménez Díaz.
Caso clínicoEl paciente es un varón de 19 meses de edad, nacido de padres sanos, no consanguíneos y sin antecedentes familiares de epilepsia. En los primeros 15 días de vida presentó crisis epilépticas tónicas, clónicas focales y oculógiras. A los 45 días de edad, el electroencefalograma mostró un trazado de brote-supresión indicativo de SO. El examen físico no reveló dismorfias y la resonancia magnética y los estudios metabólicos no aportaron resultados positivos. A los 5 meses de edad inició espasmos. El tratamiento con vigabatrina y corticoides fue efectivo, aunque el paciente desarrolló posteriormente crisis parciales complejas refractarias. Actualmente, presenta un grave retraso psicomotor, con ausencia de soporte cefálico, ausencia de lenguaje, escasa fijación visual e hipotonía generalizada.
MétodosEl ADN genómico se obtuvo a partir de linfocitos de sangre periférica usando protocolos estandarizados. La secuenciación del gen STXBP1 se realizó en ambas direcciones en los fragmentos amplificados —mediante la reacción en cadena de la polimerasa— procedentes del ADN genómico, utilizando un kit de secuenciación (Life Technologies, Carlsbad, EE. UU.) y un secuenciador ABI 3130 (Life Technologies) con cebadores específicos para el gen STXBP1. El análisis incluyó las regiones exónicas, las zonas de unión exón/intrón y las regiones reguladoras en posición 5′ y 3′ de la secuencia del gen STXBP1. La secuencia de referencia utilizada para el análisis del ARNm del gen STXBP1ha sido NM _001032221.3 (número de acceso del NCBI).
La nueva variante identificada se comprobó en 165 individuos sanos.
El ADNc se obtuvo mediante la reacción de transcripción inversa en cadena de la polimerasa del ARNm extraído de sangre total, utilizando un cebador oligo (dT) y el sistema de transcripción inversa ImProm-II (Promega, Fitchburg, EE. UU.). El ADNc fue posteriormente amplificado con cebadores específicos (5’-GAAGTCACCCGGTCTCTGAA-3’ [directo] y 5’-CACCGTGAGAGCTGGTAGGT-3’ [reverso]) para obtener un fragmento que incluyera del exón 11 al exón 16 del gen STXBP1.
La mutación se analizó con el programa Human Splicing Finder (http://www.umd.be/HSF/), una herramienta bioinformática que se utiliza para predecir el efecto de las variantes en zonas implicadas en el procesamiento del ARNm6.
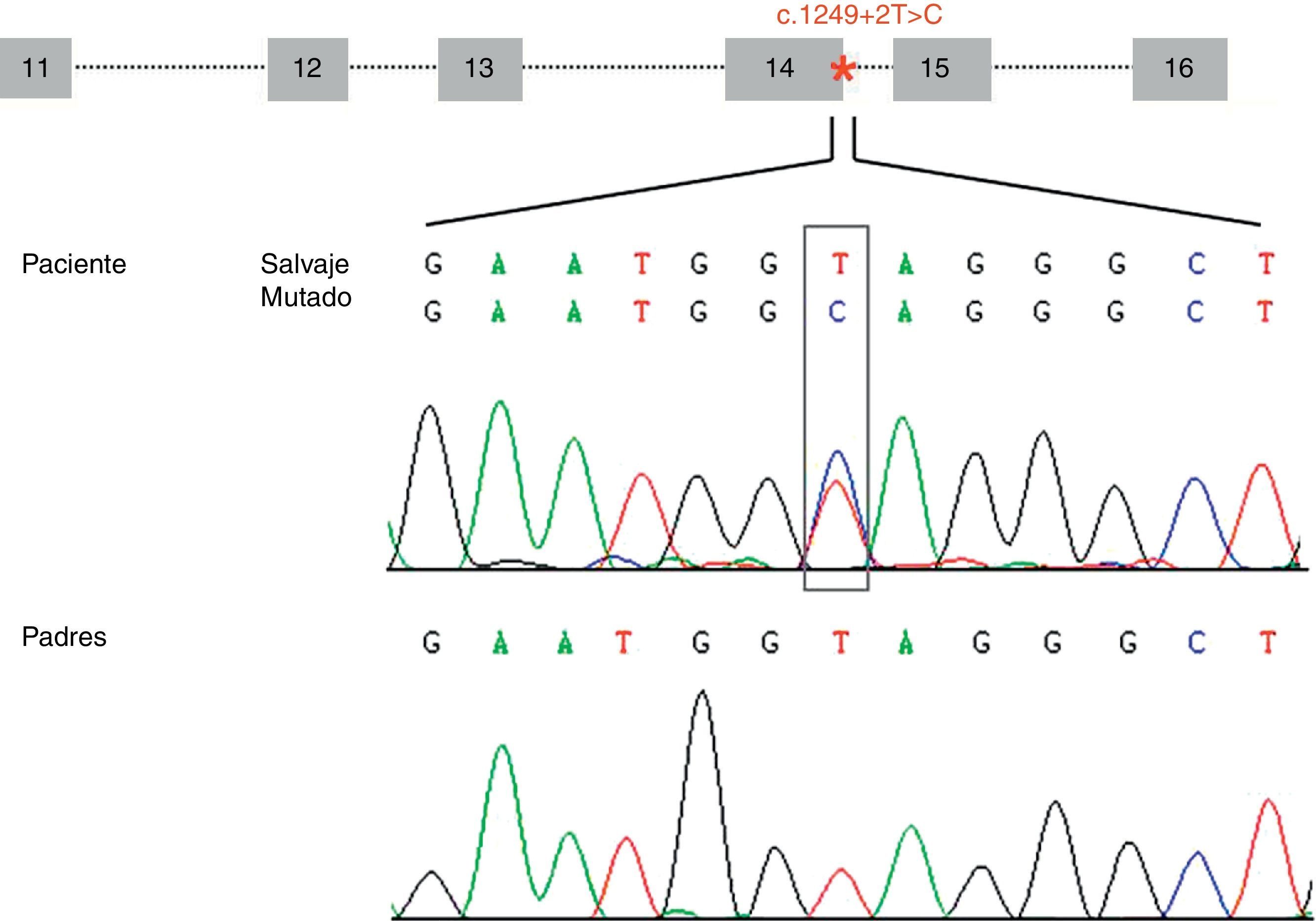
ResultadosHemos identificado una nueva mutación en heterocigosis en el gen STXBP1 (c.1249+2T>C) localizada en el sitio donador implicado en el procesamiento del ARNm del intrón 14. La mutación no se ha encontrado en los progenitores (fig. 1) y tampoco en los 165 individuos controles analizados. El análisis de esta variante con el programa bioinformático de predicción sugiere que se produce una pérdida de afinidad de la maquinaria implicada en el procesamiento del ARNm por el sitio donador que tiene la mutación (ΔCV=–31,94). No se predice activación de sitios crípticos.
 del gen STXBP1 que contiene la mutación c.1249+2T>C (localizada en el intrón 14 e indicada con un asterisco). Los electroferogramas muestran la mutación c.1249+2T>C identificada en el paciente y la correspondiente secuencia en los progenitores, sugiriendo que es una mutación de novo.")
Representación esquemática de la estructura genómica de la zona (exones del 11 al 16) del gen STXBP1 que contiene la mutación c.1249+2T>C (localizada en el intrón 14 e indicada con un asterisco). Los electroferogramas muestran la mutación c.1249+2T>C identificada en el paciente y la correspondiente secuencia en los progenitores, sugiriendo que es una mutación de novo.
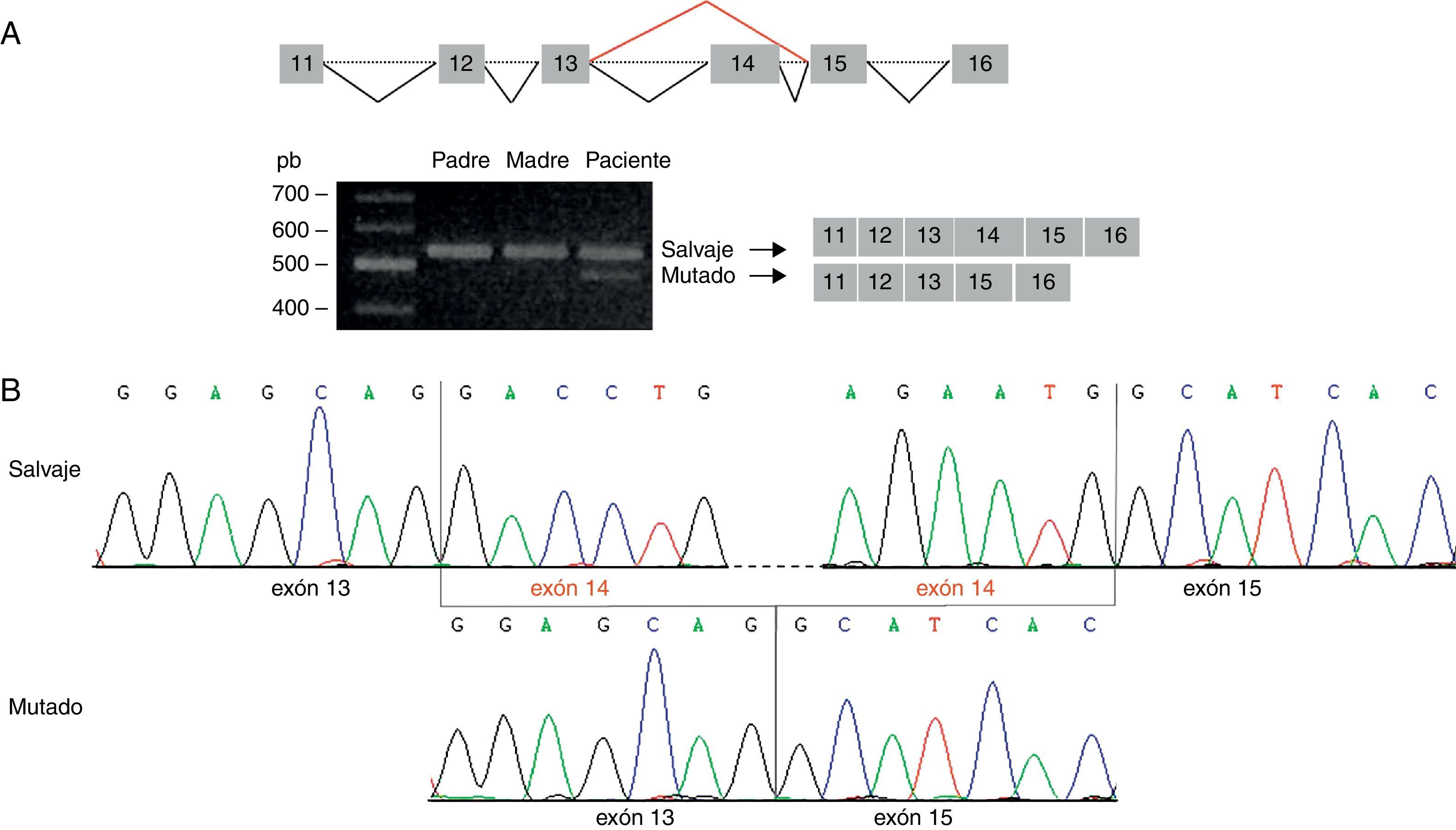
Esta predicción se confirmó mediante amplificación de la región del ADNc que contiene la variante identificada. Se visualizó una única banda de 559 pb (amplicón correspondiente al transcrito salvaje) en el ADNc de los padres, y 2 bandas en el ADNc del paciente: la banda de 559 pb y una banda de 420 pb (amplicón correspondiente al transcrito mutado) (fig. 2A). El análisis mediante secuenciación directa reveló que el amplicón de 420 pb carece del exón 14, originándose un nuevo codón de parada en la posición 424 de la secuencia proteica (G417AfsX7) (fig. 2B).
 La amplificación de la región del ADNc correspondiente a los exones 11-16 del gen STXBP1 evidenció 2 tamaños de amplicón en el paciente: uno de 559 pb, visualizado también en los progenitores, y otro de 420 pb, correspondiente al amplicón del transcrito mutado. B) La secuenciación del amplicón de 420 pb confirma la pérdida del exón 14 del gen STXBP1.")
Análisis de la mutación c.1249+2T>C por secuenciación del ADNc. A) La amplificación de la región del ADNc correspondiente a los exones 11-16 del gen STXBP1 evidenció 2 tamaños de amplicón en el paciente: uno de 559 pb, visualizado también en los progenitores, y otro de 420 pb, correspondiente al amplicón del transcrito mutado. B) La secuenciación del amplicón de 420 pb confirma la pérdida del exón 14 del gen STXBP1.
En este trabajo se ha identificado una nueva mutación en el gen STXBP1 en un paciente con diagnóstico de SO sin lesión estructural cerebral subyacente.
Desde el año 2008 se han descrito mutaciones en el gen STXBP1 en pacientes con SO, síndrome de West y EEIP, observándose en aproximadamente el 22% de los casos de SO sin lesión cerebral2,3,7.
El gen STXBP1 está localizado en el cromosoma 9q34.11, contiene 20 exones y codifica para la proteína de unión a sintaxina 1 (STXBP1). La proteína STXBP1 regula el acoplamiento de las vesículas sinápticas, así como la liberación de los neurotransmisores mediante la unión específica a sintaxina 1A (STX1A), cambiando su conformación y regulando con ello al complejo SNARE5.
La nueva mutación identificada, c.1249+2T>C, produce la pérdida del sitio donador implicado en el procesamiento del ARNm del intrón 14. A nivel de la proteína, las consecuencias implican la pérdida completa del dominio 3b y parte del dominio 2 de STXBP1. Los dominios 1 y 3a forman la cavidad central que proporciona la superficie de unión a STX1A, un paso esencial para la formación del complejo SNARE y la posterior liberación de los neurotransmisores8. Por lo tanto, la mutación identificada no debería afectar a la unión de STXBP1 con STX1A.
Se han descrito mutaciones en el mismo dominio funcional de STXBP19, indicando la patogenicidad de esta nueva mutación. Por otra parte, estudios realizados con STXBP1 ortóloga de Caenorhabditis elegans indican que cambios en los residuos aminoacídicos anteriores a la posición p.Y402 podrían afectar al proceso de fusión de las vesículas sinápticas. El transcrito mutado probablemente sea degradado a través del mecanismo NMD, un mecanismo de degradación del ARNm mediado por mutaciones terminadoras, dando lugar a haploinsuficiencia de STXBP1, como se ha descrito en otros estudios realizados con mutaciones similares10.
Esta nueva mutación en el gen STXBP1, identificada en un paciente con SO sin lesión cerebral estructural subyacente, amplía el espectro mutacional asociado a este devastador síndrome epiléptico.
Aprobación éticaCada individuo participante o representante legal ha firmado un consentimiento informado aprobado por el Comité Ético de la Fundación Jiménez Díaz.
FinanciaciónEste trabajo ha sido financiado por el Ministerio de Economía y Competitividad (SAF2010-18586 and EUI-EURC-2011-4325). Laura Ortega Moreno tiene una beca de investigación de la Fundación Conchita Rábago de Jiménez Díaz.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este trabajo fue presentado como póster en la sección de Enfermedades Neurodegenerativas del Congreso Europeo de Genética Humana (París) en 2013.

