






To report the clinical manifestations and therapeutic schemes established in three patients with cutaneous polyarteritis nodosa, as well as to describe the pathophysiology, clinical features, diagnostic criteria, and therapeutic options.
MethodsA literature review was performed using Google scholar and PubMed and MeSH terms. There was no limit on the publication date or language for the selection of the articles.
ResultsCutaneous polyarteritis nodosa is an uncommon small and medium-sized vessel vasculitis, and is rarely reported in Colombia. Although its pathophysiology is unknown, it is considered to be mediated by the deposition of immunocomplexes in the walls of blood vessels. It can be triggered by infectious agents and be associated with autoimmune diseases. The cutaneous manifestations mainly include subcutaneous painful nodules, livedo reticularis, and ulcers. Myalgia, arthralgia, peripheral neuropathy, and fever can also be present. It represents a diagnostic challenge. Treatment is not standardized and is guided according to the severity.
ConclusionsCutaneous polyarteritis nodosa is a rare entity, with a wide variety of cutaneous manifestations. There is still no specific diagnostic test. Its diagnosis represents a challenge for the dermatologist, and requires multidisciplinary management, in which the rheumatologist plays a fundamental role.
Reportar las diferentes manifestaciones clínicas y los esquemas terapéuticos instaurados en 3 pacientes con poliarteritis nudosa cutánea. Así mismo, describir la fisiopatología, el cuadro clínico, los criterios diagnósticos y las opciones terapéuticas.
MétodosSe realizó una búsqueda de la literatura en Google Académico y PubMed utilizando términos MeSH. En la selección de la bibliografía, la búsqueda no se limitó por fecha de publicación ni por idioma, debido al escaso número de reportes.
ResultadosLa poliarteritis nudosa cutánea es una vasculitis de pequeño y mediano vaso, infrecuente, poco reportada en Colombia. Su fisiopatología no es del todo conocida, se considera mediada por el depósito de inmunocomplejos en las paredes de vasos sanguíneos; puede ser desencadenada por agentes infecciosos y asociarse a patologías autoinmunes. Las manifestaciones cutáneas incluyen principalmente nódulos subcutáneos dolorosos, livedo reticular y úlceras. A nivel extracutáneo podrían encontrarse mialgias, artralgias, neuropatía periférica y fiebre. El diagnóstico representa un desafío. El tratamiento no está estandarizado y es guiado según la severidad de la patología.
ConclusiónLa poliarteritis nudosa cutánea es una entidad poco frecuente, con una amplia variedad de manifestaciones cutáneas. Hasta el momento, no contamos con una prueba diagnóstica específica. Su diagnóstico representa un reto para el dermatólogo y requiere un manejo multidisciplinario, en el cual el reumatólogo desempeña un papel fundamental.
Polyarteritis nodosa (PAN) is a systemic necrotizing vasculitis that affects small and medium-caliber arteries, mainly in the kidney, the peripheral nervous system and the skin, without lung involvement, according to the classical clinical records.1–3 It is considered a diagnosis of exclusion against other types of medium and small-caliber vasculitis and is characterized by presenting negative anti-neutrophil cytoplasmic antibodies (ANCA).4 PAN can compromise the skin in an isolated way, case in which it is called cutaneous PAN (cPAN), a clinical variant of infrequent presentation and etiology that has not yet been elucidated.1 cPAN represents between 4 and 10% of all cases of PAN.1,5 Below are reported three confirmed cases of the disease that posed a diagnostic challenge for the medical staff in charge and required different therapeutic schemes due to their variable clinical course and severity. Finally, a brief review of this entity is made.
Clinical casesCase 1A 34-year-old woman, unemployed publicist, who consulted for erythematous nodules, subintrant and painful on palpation, located on the thorax, back and lower limbs for three years. The patient described the pain as lancinating, disabling, usually of 8/10 in the visual analogue scale. On the review by systems, she referred intermittent arthralgia of low intensity in the elbows, knees and ankles, predominantly in the evening, without deformity or stiffness, as well as myalgia in the proximal forearms, the distal thighs and in the legs at the level of the gastrocnemius muscles, which were accentuated with palpation, without associated weakness in the proximal or distal segments.
Within the personal antecedents, the patient reported that 2 years ago she has received extra-institutional treatment with prednisolone and methotrexate (dose and frequency not remembered by the patient nor recorded in the medical history), as well as cyclophosphamide 6g intravenously (IV) in total, considering a diagnostic impression of non-specific inflammatory myopathy, without improvement of the lesions or related symptoms.
In addition to the skin lesions (Fig. 1), communication of both nostrils through the nasal septum, in its cartilaginous portion, painful on palpation, compatible with anterior septal perforation was evident in the physical examination. Due to this clinical finding, extension studies were carried out that allowed to rule out granulomatosis with polyangiitis and other systemic primary vasculitis, based on imaging studies of the lung and of the renal function, in addition to urinalysis and erythrocyte morphology in occasional urine sample, excluding microscopic hematuria of glomerular origin.

The following paraclinical tests were requested: Elisa for human immunodeficiency virus (HIV), hepatotropic viruses, cold agglutinins, antinuclear antibodies (ANA), antibodies against extractable nuclear antigens (anti-ENA), ANCA, rheumatoid factor (RF), anti-cyclic citrullinated peptide antibodies (anti-CCP), as well as antiphospholipid antibodies (lupus anticoagulant, anticardiolipin IgM and IgG, anti-B2-glycoprotein IgM and IgG), which were negative. Likewise, the patient denied in the anamnesis recent facial trauma (which would explain a reabsorption of septal hematoma) or consumption of inhaled alkaloids.
Subsequently, 6 bacilloscopies of the lymph were taken, which were negative for Mycobacterium leprae, and a skin biopsy in which granulomas or diffuse dermal infiltrate by histiocytes, neural or granulomatous involvement were not evidenced, with negative Fite-Faraco staining; likewise, leishmaniasis was ruled out as an associated differential diagnosis. The skin biopsy documented mixed panniculitis, predominantly septal, with medium- and small-vessel vasculitis.
The studies of electromyography and conduction velocity were negative for mononeuritis multiplex or variants of symmetric polyneuropathy. cPAN was estimated as a diagnosis of exclusion, which is why prednisolone was started at 1mg/kg/day in a progressive titration scheme for 4 weeks, without achieving clinical improvement.
Given the persistence of the lesions, the patient was referred to Rheumatology and continued with prednisolone 25mg/day, starting methotrexate at 15mg/week. Due to the poor response and poor control of pain, she received 2 additional doses of cyclophosphamide (500mg IV each, every 15 days). However, new lesions continued to appear on extensor surfaces, and for this reason it was decided to start infliximab, at a dose of 5mg/kg. Until now, there is evidence of having completed 3 doses in total (weeks 0, 2 and 6), with a decrease in the intensity of pain and in the size of the cutaneous lesions; it is still pending to continue the same agreed dose every 8 weeks, which was proposed as maintenance phase.
As for the anterior septal perforation, there is no clarity about the time of appearance and the precise cause of it. To date, the following etiologies have been ruled out: polyangiitis with granulomatosis, relapsing polychondritis, occasional use of inhaled cocaine, resolved septal hematoma, mucosal leishmaniasis, and Hansen's disease. Currently, she is undergoing outpatient follow-up by Otorhinolaryngology.
Case 2A 58-year-old woman, a housewife, with no significant personal or family history, who consulted due to a clinical picture of 8 years of evolution of painful erythematous nodules on the lower limbs, which became ulcerated in the last 3 months, associated with purulent discharge and intense lancinating pain. She had received treatment with topical antibiotics (fusidic acid, mupirocin) and multiple dressings with silver sulfadiazine, without clinical improvement. She also reported myalgia in the lower limbs, without weakness, arthralgia/arthritis or signs of joint effusion, without rigidity or associated motor deficit.
On physical examination, she presented generalized purpuric lesions with a reticular configuration (Fig. 2A and B), on the anterior region of the legs, ankles, and the dorsum of the feet, ulcers with well-defined regular borders, and others with confluent star-shaped edges, as well as areas of white atrophy on the periphery, painful, which limited walking. The initial diagnostic impression was classic PAN.

A skin biopsy was performed, which reported a small and medium-vessel vasculitis, without formation of granulomas. Perivascular deposits of IgM, C1q and C3 were observed in the direct immunofluorescence.
At the same time, the studies to rule out autoimmunity (ANA, anti-ENA, ANCA, RF, anti-CCP, lupus anticoagulant, anticardiolipin IgM and IgG, anti-B2-glycoprotein IgM and IgG) were negative. Likewise, systemic involvement of pulmonary, renal, gastrointestinal, and central and peripheral nervous systems type was ruled out. Given the negativity of the autoimmunity profile, the diagnosis of systemic lupus erythematosus was ruled out and the diagnosis of cPAN was determined by exclusion.
Immunosuppressive treatment with prednisolone 1mg/kg/day orally was prescribed, in addition to ampicillin/sulbactam, due to clinical findings indicative of bacterial superinfection. The patient improved progressively during hospitalization and was discharged with prednisolone in a conventional reduction scheme, until she was left with a maintenance dose of 7.5mg/day and an order for a new evaluation by Rheumatology. The clinical follow-up was lost due to sociodemographic issues, without knowing her evolution to date.
Case 3A 27-year-old man, photographer, who attended a medical consultation for a 3-month history of ulcerative lesions on the feet and plantar region, preceded by acral cyanosis intensified by low temperatures.
As personal antecedents, he referred juvenile idiopathic arthritis, diagnosed when he was 5 years old, which required treatment with methotrexate and from the age of 18 with etanercept for 2 years (at unknown doses and schemes) and which he discontinued at the age of 20 on his own decision. In addition, diagnosis of HIV infection since the age of 25, at the time classified as stage 2, with 100% adherence to antiretroviral therapy (efavirenz, emtricitabine, and tenofovir) and adequate immunovirological status (undetectable viral load, CD4+ lymphocyte count greater than 500 cells/mm3), with no evidence of opportunistic infections in the clinical history.
In the review by systems, he reported hypoesthesia in both feet with a "stocking" pattern, without arthralgia or joint effusion, with a secondary antalgic gait.
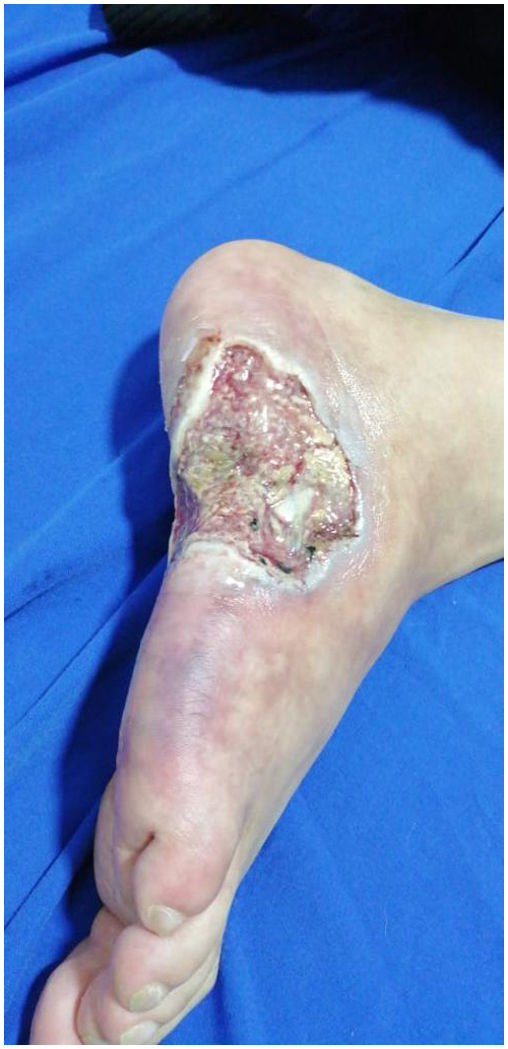
On physical examination, he presented reticulated violaceous macules of irregular and incomplete distribution on the dorsum and lateral aspect of the right foot; there was an ulcer of 6×4cm on the lateral aspect of the foot, with macerated and irregular edges, painful on palpation, with a clean base, with exposure of tendons and some erythematous and violaceous perilesional nodules smaller than 1,5cm (Fig. 3), without arthritis, dactylitis or enthesitis, nor low back pain with inflammatory characteristics. Palpable distal pulses in lower extremities (femoral, pedal, posterior tibial, popliteal), without signs of critical ischemia.

Due to the acceptable immunological control at the time of consultation, along with the exclusion of related vascular disorders, pyoderma gangrenosum and vasculitis in the setting of retrovirus infection, both of infectious etiology (vasculitis associated with cytomegalovirus [CMV], Mycobacterium tuberculosis [M. tuberculosis] and hepatitis B [HBV] and C [HCV] viruses, as well as the HIV itself, usually related with deposits of IgA) and associated with immune complexes (hypersensitivity vasculitis and angiocentric immunoproliferative vasculitis) were considered among the differential diagnoses. Complementary paraclinical tests were performed: cold agglutinins, renal function, electrolytes, as well as studies to evaluate autoimmunity (ANA, anti-ENA, ANCA, RF, anti-CCP), and antiphospholipid antibodies (lupus anticoagulant, anticardiolipin IgM and IgG, anti B2-glycoprotein IgM and IgG), which were negative.
Likewise, a computed tomography (CT) of the thorax and abdomen was carried out, simple and with contrast, without evidence of significant findings; it was not complemented with angiographic evaluation of abdominal vessels. Finally, the complementary electrophysiological studies ruled out peripheral nerve lesions.
A skin biopsy was performed, which documented small and medium-vessel vasculitis and acute abscessed inflammation, without formation of granulomas, with negative staining for fungi and mycobacteria (Fig. 4).
. Hematoxylin and eosin, 10×. Epidermis with compact orthokeratosis (*). In the deep dermis, close to the subcutaneous cell tissue, vasculitis of a medium caliber vessel with leukocytoclastia and hyperproliferation of the intima layer, without intraluminal thrombi is observed, (arrow).")
Skin biopsy (patient number 3). Hematoxylin and eosin, 10×. Epidermis with compact orthokeratosis (*). In the deep dermis, close to the subcutaneous cell tissue, vasculitis of a medium caliber vessel with leukocytoclastia and hyperproliferation of the intima layer, without intraluminal thrombi is observed, (arrow).
The juvenile idiopathic arthritis was considered to be in remission, due to the absence of clinical symptoms or signs of activity, without medications that could generate these clinical findings. cPAN was considered as the most probable diagnostic option, ruling out as the main differential diagnosis PAN-like associated with HIV infection, a secondary vasculitis that may occur with any CD4+ lymphocyte count.
Thereafter, treatment with prednisolone 1mg/kg/day (50mg/day) and azathioprine 50mg/day orally was indicated, with notable clinical improvement 4 weeks after starting the treatment. Subsequently, the dose of the steroid was decreased by conventional scheme up to 20mg/day and the azathioprine dose already indicated was continued. The patient, who is still being followed-up by the dermatology service, has managed to resolve the antalgic gait and shows progress in the healing process of his lesions
DiscussionPAN was the first systemic vasculitis described in the medical literature. By 1866, Kussmaul and Maier characterized this condition based on the anatomopathological findings of a necrotizing vasculitis of medium and small arteries (excluding arterioles, capillaries and venules).1,2 There are 3 clinical variants described for this entity: the classic idiopathic form, the one associated with HBV infection, and the cPAN. The latter, described by Lindberg in 1931 as a vasculitis of medium-caliber blood vessels limited to the skin,3,5–7 can evolve into classic PAN, in which case the prognosis and treatment would change. According to the latest Chapel-Hill consensus of 2012, this variant is reclassified as a single-organ arteritis (in this case, the skin).6–8
There are no reports of prevalence or incidence of cPAN due to its infrequent presentation. A study that included 79 cases of cPAN found a female:male ratio of 1.7:1.4,6 cPAN mainly affects women over 40 years of age, with a peak between 50 and 59 years of age.5,8,9 In the registry of Nakamura et al.,10 with 22 patients aged between 17–77 years, 86% were women. The foregoing correlates with our report, in which 2 of the 3 cases were women, both over 40 years of age. The reports of documented cases in Colombia to date are included in Table 1.11–14
Registries of cutaneous PAN (cPAN) in Colombia.
| Iglesias et al.11 | Olmos et al.12 | Quintana et al.13 | Cárdenas et al.14 | Ordóñez et al. (current) | |
|---|---|---|---|---|---|
| Case series | Case report | Case report | Case report | Case report | |
| Distribution by gender (number of cases) | 4 cases: women (3), man (1) | Man (1) | Woman (1) | Woman (1) | 3 cases: women (2), man (1) |
| Mean age | 18−44 years | 19 years | 36 years | 58 years | 27−58 years |
| Clinical manifestations | |||||
| Erythematous nodules (location) | + | + | + | + | + |
| (upper and lower limbs) | (upper and lower limbs) | (thigh, right breast, forearms, scalp and upper third of the right leg) | (lower limbs) | (thorax, back [1] and upper and lower limbs) | |
| Fever | + | + | – | – | – |
| Myalgia | + | + | – | – | + |
| (upper and lower limbs) (2) | |||||
| Arthralgia (location) | + | + | – | – | + |
| (knees and ankles) | (elbows, knees and ankles) (2) | ||||
| Livedo reticular | – | + | – | + | + |
| (neck, sacral region and extremities) | (lower limbs) | (lower limbs) (2) | |||
| Ulcers | + | – | – | + | + |
| (upper and lower limbs) | (lower limbs) | (lower limbs) (2) | |||
| Pathological findings (skin biopsy) (number of cases) | Leukocytoclastic vasculitis of medium-caliber arteries and arterioles with fibrinoid necrosis | Leukocytoclastic vasculitis of medium-caliber arteries, with thrombosis and total occlusion of their lumen due to fibrin deposition | Leukocytoclastic vasculitis of medium-caliber arteries with fibrinoid necrosis | Leukocytoclastic vasculitis of medium-caliber arteries and arterioles with fibrinoid necrosis | Mixed panniculitis with septal predominance (1), medium- and small-vessel vasculitis, without granulomas (3) |
| Direct immunofluorescence | Deposit of Ig M, G and C3 in the wall of the vessels | – | – | – | Deposits of IgM, C1q and C3 in the wall of the vessels (1) |
| Treatment (number of cases) | Prednisone 30−60 mg | Prednisone 0.5mg/kg/day | Prednisone 15−30mg/day | Prednisolone (do not describe doses) | Prednisolone 1mg/kg/day (2) then 20−25mg/day (2) |
| Azathioprine 100mg/day | Azathioprine 100mg/day | Cyclophosphamide 500mg IV monthly, for 2 years | Methotrexate 15mg/weekly (1) | ||
| Cyclophosphamide 100mg orally daily+prednisone | Colchicine 1mg/day | Azathioprine | |||
| Cyclophosphamide, 500mg IV monthly×2 doses | Two doses of cyclophosphamide 500mg IV (1) | ||||
| Infliximab 5mg/kg/dose weeks 1, 2, 6 and every 8 weeks |
The etiology of cPAN is unknown,1–5 it is believed to be a disorder mediated by immune complexes, with deposition of C3 and IgM on the walls of the blood vessels.8–10 Infectious agents such as hepatotropic viruses (B and C) have been associated with its onset or recurrence,2,10,15 therefore, it is mandatory to perform serology for these viruses in patients in whom this clinical condition is suspected, as was done in the cases presented.
Criado et al.16 reported M. tuberculosis as the most commonly associated infectious agent, followed by CMV. The most accepted mechanism is the stimulation of autoreactive T and B lymphocytes, since the Ziehl-Neelsen stainings have been negative to detect the presence of mycobacteria in skin biopsies.
In developing countries, cPAN has been associated with HIV infection.15,17 Even though HIV per se can trigger a vasculitis which is not exclusively of small size,15 in case 3 the patient met clinical and histological characteristics of cPAN, having ruled out another type of vasculitis, for which it was considered an immunologic epiphenomenon associated with the underlying viral infectious disease.
Among the non-infectious causes, it has been described and association with myasthenia gravis, inflammatory bowel disease and autoimmune hepatitis, while its recurrence has been related recently as a herald of neoplastic processes of hematolymphoid type (mainly myelodysplastic syndrome). Drugs such as penicillin and tetracyclines have been reported as triggering agents in the development of cPAN, with clinical improvement after their immediate suspension.17,18
On the other hand, the presence of IgM antibodies against the phosphatidylserine-prothrombin complex (anti-PS/PT) has been demonstrated in up to 81.3% of the patients with cPAN, with a positive correlation between anti-PS/PT levels and C-reactive protein (CRP),2,3 which supports the theory of prothrombin binding to apoptotic endothelial cells, triggering an immune response mediated by reactive T and B lymphocytes.5,10,19 In addition, in cPAN there is an activation of the classical complement pathway, apparently secondary to the deposition of IgM and IgG immunoglobulins.
The cutaneous manifestations of classic PAN and cPAN can be very similar. Classic PAN involves painful subcutaneous nodules in the legs (97% of cases), head and neck (39%), arms (33%) and trunk (8%).7,19 In a retrospective registry of 79 patients with cPAN published by Daoud et al.20 it was reported that 80% had painful nodules in the lower limbs with edema; 56% presented livedo reticularis, 49% ulcers, and 10% indurated plaques. In addition, it was described that in up to 31.3% of the patients there was evidence of hemorrhagic lesions, from petechiae to extensive purpura. To date, there is no evidence of anterior septal perforation as an associated clinical manifestation of cPAN, as described in case 1, in which the most frequent causes were ruled out.
Nakamura et al.21 reported 22 patients in whom subcutaneous nodules were found in the lower limbs (100%) and in the upper limbs (16%); livedo reticularis in the lower limbs (80%), the forearms (10%) and the back (10%); purpura in the lower limbs (100%) and ulcers in the lower limbs (100%). The nodules were present in the 3 cases described and 2 of them presented livedo racemosa around an ulcer, which demonstrates the variability in location and morphology of the lesions. White atrophy lesions, as in case 3, have been reported in up to 27.3%, and the referred pain in the area or the nodules and ulcers is usually intense.17,21
The 3 cases described here referred severe lancinating pain preceded by myalgia, mainly in case 1, which led us to think of a neuropathic involvement in the context of leprosy22 and erythema nodosum leprosum, or even in the course of other ANCA-associated vasculitides, such as granulomatosis with polyangiitis or microscopic polyangiitis. The absence of visceral involvement is what makes it possible to differentiate cPAN from the classic variety, so that in the face of characteristic cutaneous lesions, systemic involvement must always be ruled out.
Likewise, the extracutaneous manifestations that could accompany cPAN should be kept in mind, such as fever (18%), weight loss (5%), peripheral neuropathy (32%), myalgia (27%) and arthralgia (18%), whose presence would allow lowering the diagnostic threshold and intervening in the course of the disease in a more timely manner.6,7,19
To date, there are no specific diagnostic tests for cPAN. The diagnosis is defined by a comprehensive correlation between the clinical symptoms and signs, the histopathological findings and the complementary laboratory tests to rule out other entities. Table 2 describes a proposal for clinical diagnosis accepted by the Ministry of Health, Labour and Welfare of Japan based on the study of 22 cases of cPAN.21
Diagnostic criteria proposed for cutaneous PAN (cPAN).
| Cutaneous manifestations | Subcutaneous nodules, livedo reticularis, purpura, ulcers |
|---|---|
| Histopathological findings | Fibrinoid necrotizing vasculitis of small and medium caliber arteries |
| Exclusion criteriaa | (1) Fever (=38°C,=2 weeks), weight loss (6kg or more in 6 months) |
| (2) Arterial hypertension | |
| (3) Rapidly progressive renal failure, renal infarction | |
| (4) Cerebral hemorrhage, cerebral infarction | |
| (5) Myocardial infarction, ischemic heart disease, pericarditis, heart failure | |
| (6) Pleuritis | |
| (7) Intestinal bleeding, intestinal infarction | |
| (8) Peripheral neuropathy besides the affected cutaneous lesion | |
| (9) Arthralgia (arthritis) or myalgia (myositis) besides the cutaneous lesion | |
| (10) Abnormal angiography (multiple microaneurysm, stenosis, and obliteration) | |
| Decision | Correlation between clinical and histopathological findings and absence of all exclusion criteria |
Taken from Nakamura et al.21.
The Japanese, European, and Turkish series suggest to carry out a complete blood count, erythrocyte sedimentation rate, CRP, renal function, and complete liver biochemistry as initial studies, in addition to ruling out HIV, HBV, and HCV infection.23–25 The serological tests for syphilis, ANA, anti-ENA, ANCA, RF, and anti-CCP are usually negative; however, positivity for p-ANCAS has been described in the group of patients with minocycline-induced cPAN.15,23 Even, up to 43.8% may have positive lupus anticoagulant at variable titers.3,4
The histopathology is characterized by the presence of leukocytoclastic vasculitis and fibrinoid degeneration of small and medium-sized vessels in the dermis and the subcutaneous cellular tissue.7,18 Four stages that show the histopathological evolution of cPAN have been described: the first or degenerative, characterized by deposition of fibrinoid material and destruction of the elastic lamina; the second, or inflammatory, with a neutrophilic infiltrate and some eosinophils with a perivascular distribution; the third or granulation, consisting of a lymphohistiocytic infiltrate, with proliferation of the intima and thrombosis of the artery; and the fourth or final stage, with perivascular fibroblastic proliferation.1–3,21
Gupta et al.24 described the inflammation of the adipose panicle in cPAN, with a neutrophilic infiltrate and some perivascular eosinophils located in the fat lobule, which was the cause of ischemia and ulceration. The 3 cases presented small- and medium-vessel vasculitis, in addition to panniculitis, as described in the patient of case 1.
As for direct immunofluorescence, perivascular deposits of IgM or complement could be observed, which supports the theory of immune complexes. In case 2, deposition of C1q was also observed, which obliged to rule out differential diagnoses such as systemic lupus erythematosus, since these findings are typical of the so-called lupus band.
Exacerbations are frequent and can last between 2 and 8 weeks. The following have been described as factors associated with a worse prognosis (recurrence in the next 2 years of follow-up): multiple ulcers at the beginning and elevated acute phase reactants before starting treatment.25–27
In turn, Chen28 reported that patients with myalgia, arthralgia, and symptoms associated with peripheral nerve involvement (dysesthesias, paresthesias) had more frequent and severe relapses.26 Progression from cPAN to classic PAN has been reported in up to 15–20% of the patients, therefore, progression should be ruled out during each relapse.8–10 It has been described a greater systemic progression in patients with positive RF and ANA at high titers.29–31
Until now, there are no prospective studies or evidence-based guidelines for the treatment of cPAN. The therapeutic strategies are based on case reports, case series and literature reviews. Table 3 summarizes the treatments and doses that are described in the literature to date. The treatment depends on the clinical manifestations and the severity.29
Doses and therapeutic schemes for cutaneous PAN (cPAN).
| Therapeutic option | Severity | ||
|---|---|---|---|
| Mild | Severe | Refractory | |
| First line | Monometasone furoate 0.1% cream, clobetasol propionate 0.05% cream or ointment: 2 times a day on the lesion with occlusion for 8−12 weeks27,31,34 | Prednisolone: 30mg/day-1mg/kg/day for at least 4 weeks and then a progressive decrease until the minimum effective dose 21,30,31 | Intravenous immunoglobulin: 1−2g/kg every 4 weeks41,42 |
| NSAID: ibuprofen up to 800mg every 8h, naproxen 500mg every 12 h27,30 | Azathioprine: 2−3mg/kg/day30,37 | Infliximab: 5mg/kg/week 0, 2643,46 | |
| Colchicine: 0.5−1mg/day30 | Methotrexate: 15−22.5mg/weekly31,36 | Etanercept: 50−100mg/weekly44–46 | |
| Chloroquine: 3mg/kg/day31,38 | |||
| Hydroxychloroquine: 6mg/kg/day31,38 | |||
| Mycophenolate mofetil: 1−3g/day35 | |||
Relative rest, periodic changes of position (including Trendelenburg position), non-steroidal anti-inflammatory drugs (NSAIDs) or colchicine are indicated as first line in mild cPAN; high-potency topical steroids such as clobetasol can be added in the case of very localized or limited lesions.31–34 The use of dapsone has been reported with variable results.32 Pentoxifylline and sulfasalazine could be used as adjuvant therapy, never as monotherapy.31,34
In patients with more extensive involvement or in severe cases, oral prednisolone is the treatment of choice.30,32,33 In the 3cases presented, this was the therapy of choice, at the doses recommended by the literature. The use of azathioprine, methotrexate, antimalarials (chloroquine, hydroxychloroquine) and mycophenolate mofetil is frequently required as adjuvant immunosuppressive therapy,35,36 since these drugs allow de-escalation of steroid doses, better control of the symptoms and decrease in relapses.4,31–34,37
In patients with poor control of the disease, the use of IV cyclophosphamide in pulses, associated with the oral steroid, has shown high remission rates. In these cases, the scheme usually indicated for ANCA-associated vasculitis in the induction phase is outlined: 0.75mg×m2 of body surface area IV every 4 weeks, without exceeding 1g per infusion for 6 months. In patients with impaired renal function, the dose is modified as follows: 0.5mg×m2 of body surface area; likewise, in patients over 65 years of age the dose per pulse will be 500mg IV every 15 days, for a total of 6 doses.38–40
IV immunoglobulin is the last therapeutic line in cases of refractory disease,41,42 as well as agents directed against the tumor necrosis factor alpha (TNF-α), particularly infliximab or etanercept.43,44 Other therapeutic options, such as tamoxifen, hyperbaric oxygen, warfarin, and granulocyte colony-stimulating factor (G-CSF), have also been reported as adjuvants, with limited clinical evidence to date.45–50
Case 1 demonstrates not only the importance of an adequate diagnostic approach, starting from the exclusion of the most frequent diagnoses associated with the lesions described, going through the multisystem involvement typical of the classic PAN until reaching circumscribed cutaneous manifestations, but also the therapeutic and prognostic implications that the cases of high recurrence or the appearance of new lesions entail, even including biological therapy within the management alternatives.
With respect to this same case, several difficulties arise in terms of diagnosis and follow-up:
The clinical picture started with diffuse subcutaneous nodules, associated with perforation of the nasal septum; the patient was previously immunosuppressed, which may mask the current presentation of the documented manifestations.
Renal biopsy, CT angiography or arteriography were not available to rule out involvement of medium-caliber abdominal vessels, so it is not possible to completely rule out small-vessel vasculitis or systemic PAN.
The response to infliximab can be seen in systemic PAN.
ConclusionscPAN is an infrequent clinical variant within the spectrum of small- and medium-vessel vasculitis. It is characterized by the presence of painful subcutaneous nodules on the legs, associated with livedo reticularis, with a high risk of ulceration. It has a benign, chronic and variable course, with high recurrence rates and association with both infectious (HIV, hepatotropic viruses, tuberculosis, leprosy) and non-infectious (drugs, connective tissue diseases) processes; in some occasions it is considered a paraneoplastic phenomenon, mainly of hematolymphoid neoplasms.
The diagnosis of cPAN is challenging, since there is no specific marker of the disease and the clinical manifestations can be overlapped with other inflammatory and infectious entities, and even related with phenomena of hypersensitivity to some drugs. According to the characteristics of the lesions and the related symptoms, among which myalgia stands out, the early indication of skin biopsy is essential to narrow down the list of possible diagnoses and reach the cPAN, after a process of clinical reasoning for exclusion.
To date, there are no treatment guidelines for cPAN. It is recommended to start with systemic steroids, associated with other immunomodulators that allow early reduction of the conventional doses. Likewise, there are anecdotal reports of response to cytostatics in induction regimens proposed for patients with ANCA-associated vasculitis, as well as the administration of biological therapy (anti-TNF-α agents, as in case 1) in patients with predictors of poor prognosis, including onset with multiple lesions and a high recurrence rate in the first year.
LimitationsThis series of 3cases has some limitations. Due to the COVID-19 pandemic, the follow-up of patients has been lost, both in person and remotely, without knowing their clinical evolution to date. Likewise, for some immunomodulatory drugs, it was not possible to document the exact dose (as in case 1) because they were indicated extra-institutionally and there was no previous clinical history.
Ethical responsibilitiesProtection of people and animals. The authors declare that no experiments were performed on human beings or animals for this research.
Data confidentiality. The authors declare that they have followed the protocols of their workplace on the publication of patient data.
Right of privacy and informed consent. The authors have obtained the informed consent of the patients mentioned in the article.
FundingThis work did not receive funding of any kind.
Conflict of interestThe authors declare that they do not have any conflict of interest.
