




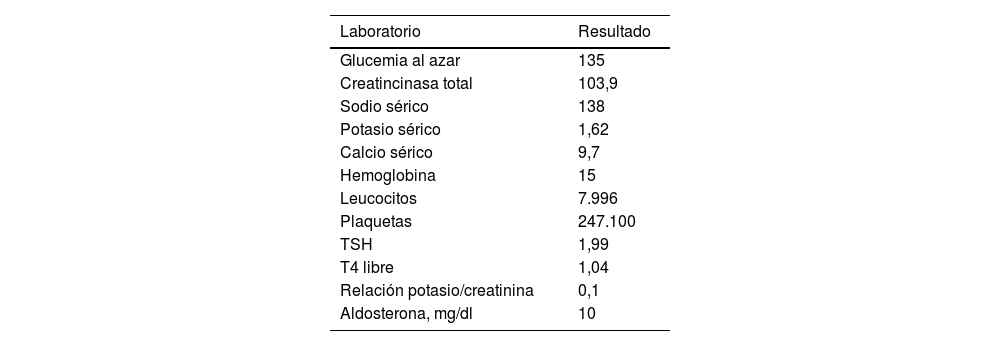
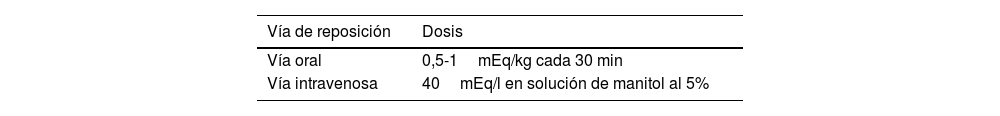
La parálisis hipopotasémica familiar es una canalopatía muscular, poco frecuente, de predominio en el sexo masculino, caracterizada por debilidad muscular generalizada asociada a hipopotasemia, con recuperación completa de síntomas posterior a la normalización de los niveles séricos de potasio; los principales factores desencadenantes son el cese del esfuerzo después del ejercicio extenuante y las cenas ricas en hidratos de carbono. A continuación presentamos el caso de un adolescente masculino con antecedente familiar de cuadro similar, quien posterior a ingesta alta de hidratos de carbono cursa con primer episodio de debilidad muscular aguda asociado a hipopotasemia severa, cuya sintomatología resuelve con la corrección del trastorno electrolítico, cumpliendo así los criterios diagnósticos de parálisis hipopotasémica familiar. La principal lección que nos deja este caso es que, si bien es una enfermedad poco común, se debe incluir su sospecha diagnóstica en un paciente con debilidad muscular.
Familial hypokalemic paralysis is a rare muscular channelopathy, predominantly in males, characterized by generalized muscle weakness associated with hypokalemia, with complete recovery of symptoms after normalization of serum potassium levels; The main triggers are cessation of effort after strenuous exercise and carbohydrate-rich dinners. Below we present the case of a male adolescent with a family history of a similar condition, who after high carbohydrate intake presents with the first episode of acute muscle weakness associated with severe hypokalemia, whose symptoms resolve with the correction of the electrolyte disorder, thus meeting the criteria. Diagnoses of familial hypokalemic paralysis. The main lesson that this case leaves us is that although it is a rare disease, its suspected diagnosis should be included in a patient with muscle weakness.
Artículo
Socios de la Asociación de Medicina Crítica y Cuidado Intensivo
![]()
Para acceder a la revista
Es necesario que lo haga desde la zona privada de la web de la AMCI, clique aquí

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
