


Primary antibody deficiencies (PADs) are a heterogeneous group of disorders, characterised by increased susceptibility to recurrent bacterial infections. Common variable immunodeficiency (CVID) is the most important PAD from the clinical point of view and selective IgA deficiency (IgAD) is the most common PAD. However, the underlying gene defect in both is still unknown. As a recent study in Europe showed an association between a single nucleotide polymorphism (SNP) of AICDA gene with PADs, this study was performed to evaluate such an association in Iranian patients.
MethodsFifty-eight patients with PAD, including 39 CVID and 19 IgAD, as well as 34 healthy volunteers, were enrolled in this study. Genotyping was done in all groups for an intronic SNP in AICDA (rs2580874), using real-time PCR genotyping assay.
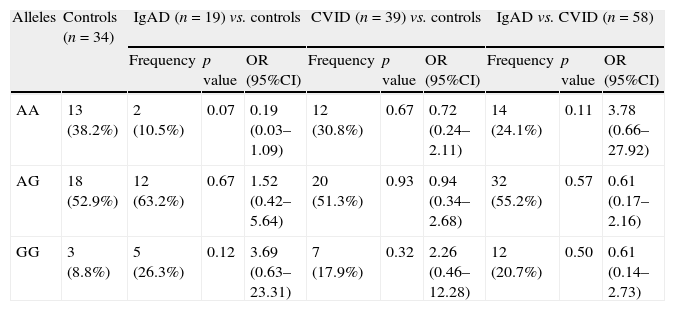
ResultsThe less frequent genotype of AICDA in IgAD patients was AA, seen in 10.5% of the patients, which was much lower than the 30.8% in CVID patients and 38.2% in the controls. However, these differences were not significant. Indeed the GG genotype in the patients with PADs was seen in 20.7%, compared to 8.8% in the controls without any significant difference.
ConclusionsThere was no significant association between the previously reported genetic variant of AICDA gene and the development of CVID or IgAD, but further multi-center studies are also needed.
Primary antibody deficiencies (PADs) are the most common form of primary immunodeficiency diseases (PIDs), and consist of a number of diseases, ranging from severe reduction of all serum immunoglobulins and decreased number of B-cells to selective immunoglobulin deficiency.1 Common variable immunodeficiency (CVID), as the most prevalent symptomatic PID, is a heterogeneous group of disorders with reduction of at least two immunoglobulin levels; some of the patients might have substantial amounts of IgM.2–5 On the other hand, selective IgA deficiency (IgAD), another heterogeneous condition, is the most common PID, resulting in reduced level of serum IgA in the presence of normal levels of other immunoglobulins.6 Although the exact pathophysiology of CVID and IgAD are still unknown, it seems that they could share a common underlying genetic defect in at least one subset of patients.6,7 In both diseases, terminal differentiation of B cells to plasma cells of the different isotypes is impaired. Some patients with IgAD also have IgG subclass deficiency and occasionally they can progress to CVID.8 Both diseases may occur in different members of the same family.7,9 Several common susceptibility haplotypes in three classes of major histocompatibility complex (MHC) have been found in CVID and IgAD.7,10 These observations suggest that genetic factors play a central role in the pathogenesis of both IgAD and CVID.11 However, neither IgAD nor CVID can be attributed solely to a simple Mendelian trait and it seems that these diseases are the result of a complex interplay between a number of genes.7 However, associations of diseases with several immunological parameters and some genetic variants, especially TNFRSF13B, have been reported.12–23
Although defective class-switch recombination (CSR) is the main basis of hyper IgM (HIGM) syndrome, it has also been proposed as a basis for CVID and IgAD.24,25 CSR is an essential process which enables B cells to differentiate to class switched B cells and produce antibodies of different isotypes. Somatic hyper mutation is another essential process for affinity maturation of antibodies produced by B cells.26,27 Class switched memory B cells are decreased in some patients with CVID and also decreased levels of SHM have been reported in subsets of these patients.28 Activation-induced cytidine deaminase (AICDA), a gene located at 12p13.31, is responsible for encoding a 198 amino-acid protein, named as AID.29,30 Mutations in this gene have been previously reported to be the most frequent aberration among patients with autosomal recessive forms of HIGM,26,30 a disease caused by defects in immunoglobulin CSR process and therefore presents with normal or elevated serum levels of IgM and low serum levels of IgG, IgA, and IgE.27,31–34 AID also has been showed to be involved in SHM process.27,29,30,35 Although the exact role of AID in CSR and SHM is not clear, two hypotheses of RNA-editing and DNA deamination have been proposed as possible mechanisms of action for AID.26,36–39 In both of them, AID exerts its action through cytidine deamination of cytidine to uracil and therefore changing the sequence of DNA or RNA.26
Lower levels of AID expression in response to TGF-β, IFN-γ and IL-10 have been reported in B cells from patients with IgAD.40 Recently, an association between rs2580874, an intronic SNP in AICDA, and IgAD and CVID has been reported.41 Therefore, AICDA defects have been proposed as the underlying cause in a subset of patients with normal IgM levels or decreased SHM who represent a new subclass of patients with HIGM.41,42 In order to evaluate the association of this SNP with IgAD/CVID, allele frequencies of this SNP were investigated in a group of patients with CVID and IgAD.
Patients and methodsStudy populationThirty-nine patients with CVID (21 male and 18 female) and nineteen patients with IgAD (10 male and 9 female), who referred to the main referral hospitals in Tehran, including Children's Medical Center, Rasoul-e-Akram Hospital, and Masih Daneshvari Hospital, were enrolled in this study as the patient group. The diagnosis of CVID and IgAD was based on standard criteria.43 Thirty-four healthy volunteers (20 male and 14 female) with the same ethnicity of the patients and without any history of severe infections and hospitalisation were also recruited as the control group to determine background population allele frequencies. They were all asymptomatic. This study was approved by the local Ethics Committee of the University. Written informed consents were taken from all the participants before sampling.
SNP genotypingFive mL of peripheral blood was collected from all the participants in EDTA-treated tubes. Genomic DNA was extracted from peripheral blood leucocytes by proteinase K phenol–chloroform extraction method. We have determined the allele frequencies for one variant in intronic region of AICDA (rs2580874), using real-time PCR allelic discrimination TaqMan genotyping assays (Applied Biosystems, Foster city, USA) and ABI 7300 Real-Time PCR system. Reactions were processed following standard protocols for Applied Biosystems.
Statistical analysisStatistical analyses were done using ABI SDS V software, version 1.4. A p-value less than 0.05 was considered statistically significant. Odds ratios (OR) and 95% confidence intervals (95% CI) for the effect of all SNPs on IgAD/CVID risk were estimated.
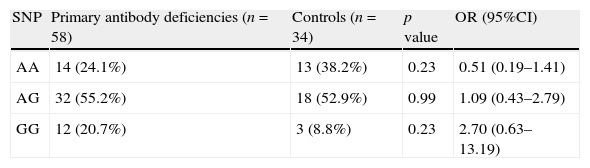
ResultsAnalysis of AICDA gene at rs2580874 showed that the less frequent genotype in IgAD patients was AA, seen in 10.5% of the patients, which was much lower than the 30.8% in CVID patients and 38.2% in the controls. However, these differences were not significant (Table 1). Fourteen of 58 antibody deficient patients had AA genotype, which was insignificantly higher than the controls (p=0.23) (Table 2).
Genotype frequencies of AICDA gene at rs2580874 in patients with CVID, IgAD, and control individuals.
| Alleles | Controls (n=34) | IgAD (n=19) vs. controls | CVID (n=39) vs. controls | IgAD vs. CVID (n=58) | ||||||
| Frequency | p value | OR (95%CI) | Frequency | p value | OR (95%CI) | Frequency | p value | OR (95%CI) | ||
| AA | 13 (38.2%) | 2 (10.5%) | 0.07 | 0.19 (0.03–1.09) | 12 (30.8%) | 0.67 | 0.72 (0.24–2.11) | 14 (24.1%) | 0.11 | 3.78 (0.66–27.92) |
| AG | 18 (52.9%) | 12 (63.2%) | 0.67 | 1.52 (0.42–5.64) | 20 (51.3%) | 0.93 | 0.94 (0.34–2.68) | 32 (55.2%) | 0.57 | 0.61 (0.17–2.16) |
| GG | 3 (8.8%) | 5 (26.3%) | 0.12 | 3.69 (0.63–23.31) | 7 (17.9%) | 0.32 | 2.26 (0.46–12.28) | 12 (20.7%) | 0.50 | 0.61 (0.14–2.73) |
Comparison of genotype frequencies of AICDA gene at rs2580874 between group of patients with primary antibody deficiencies and control group.
| SNP | Primary antibody deficiencies (n=58) | Controls (n=34) | p value | OR (95%CI) |
| AA | 14 (24.1%) | 13 (38.2%) | 0.23 | 0.51 (0.19–1.41) |
| AG | 32 (55.2%) | 18 (52.9%) | 0.99 | 1.09 (0.43–2.79) |
| GG | 12 (20.7%) | 3 (8.8%) | 0.23 | 2.70 (0.63–13.19) |
Indeed, the GG genotype in the patients with PADs was seen in 20.7%, compared to 8.8% in the controls without any significant difference (p=0.23).
There was no significant difference on either GG or AA genotype between the two groups of CVID and IgAD (p=0.23).
DiscussionIn this study, the contribution of one intronic SNP in AICDA gene was analysed toward susceptibility to IgAD and CVID, two heterogeneous group of disease which comprises two poles of hypogammaglubinaemia. In this study, we did not find any association between genotypes of this SNP with either IgAD or CVID.
In accordance to our results, no AICDA mutations have previously been reported in CVID patients.42 In a study on Danish patients no association was found between 7888 T/C in exon 4 of AICDA and CVID.42 Although decreased levels of SHM and CSR are a pathological feature in some patients with IgAD and CVID, a block in differentiation of the mature B cell into a plasma cell is supposed to play a central role in the pathogenesis of both diseases.7,42 Patients with IgAD have normal quantities of IgA-bearing B-cell precursors and the majority of CVID patients have normal numbers of IgA, IgG, and IgM-bearing B cell precursors.7 So, in the majority of patients, B cells can normally undergo the CSR process.
Previously, a study on Swedish patients with CVID and IgAD found an association between this SNP and susceptibility to IgAD and CVID.41 By finding this association, Offer et al. suggested a hypothesis that a subset of patients with IgAD and CVID are predisposed to hypogammaglubinaemia due to variations in AID and they may be a new subclass of HIGM type 2.41 Despite being in an untranslated region, they proposed that this SNP is linked to unidentified alleles, which may create splice variants or unstable mRNA or affect epigenetic modulation of AID expression.41
Several reasons can explain the discordance between our results and the Swedish study. CVID and IgAD are highly heterogeneous and defects in multiple pathways may result in these phenotypes; so, variations in AID may be a risk factor for the disease in a subset of patients, who comprise a small percentage of our study population. In addition, the frequency of alleles in SNPs is highly dependent on ethnicity. Offer et al. also evaluated a larger population, which resulted in higher statistical power to detect the associations.41
In summary, we found no association between the previously reported genetic variant in AICDA and development of CVID or IgAD. Despite our negative result, it is important to identify a possible association between CVID and IgAD and variations of genes involved in CSR, mismatch repair system and SHM. These studies may result in identification of the molecular basis of these diseases and classification of the patients into subgroups.42 Further multi-center studies on a large number of patients are needed in this regard.
Ethical disclosuresProtection of human subjects and animals subjectsThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of DataConfidentiality of Data. The authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Conflict of interestThe authors have no conflict of interest to declare.
This study was supported by a grant (90-03-30-13868) from Tehran University of Medical Sciences.
