


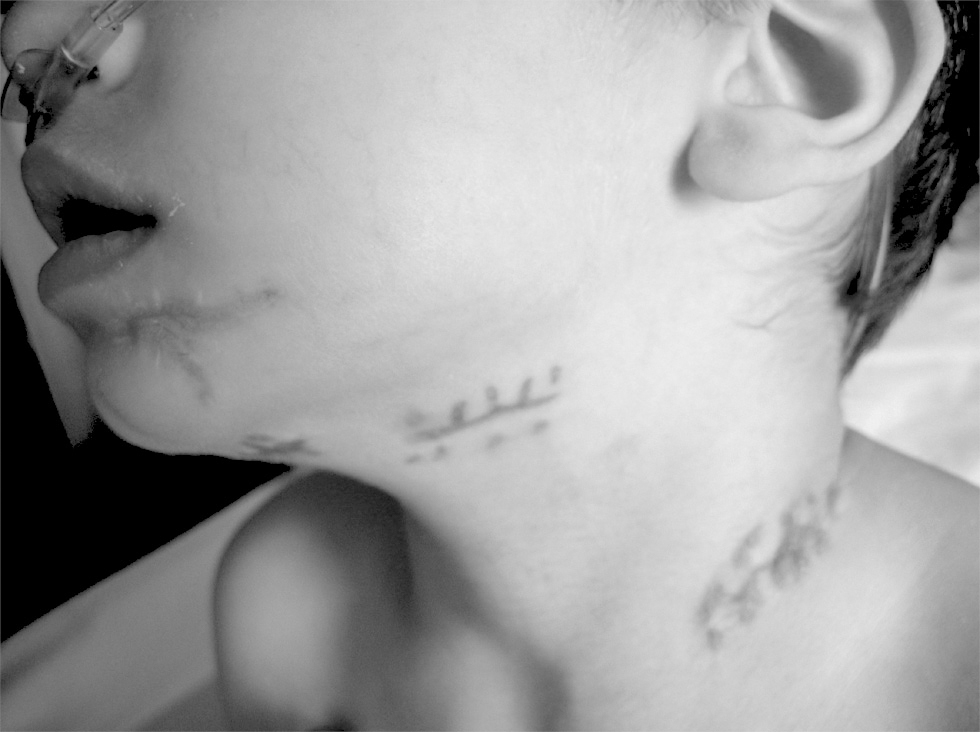
INTRODUCTION
Chronic granulomatous disease (CGD) is an uncommon primary immune deficiency (affecting 1/200,000 to 1/250,000 newborn infants) caused by a defect in phagocyte (neutrophils, monocytes, macrophages, etc.) bactericidal and fungicidal activity. These cells are unable to eliminate the phagocytosed microorganisms1-3. At molecular level, the disease is caused by different mutations of the gene that encodes for the enzyme dinucleotide phosphate oxidase (NADPH oxidase), in charge of regulating free oxygen radical production (superoxide anions, hydrogen peroxide, hydroxyl radical) for the destruction of phagocytosed microorganisms.
The NADPH oxidase system in turn is composed of different subunits. The gp91phox and p22phox subunits are located in the cell membrane, forming cytochrome b558 while the other two subunits, p47phox and p67phox, are located in the cytoplasm. The gene encoding for gp91 is found in the X chromosome, and its mutations are inherited in a recessive manner. The other genes are located in autosomes, and their mutations show a recessive autosomal hereditary pattern4-6.
The disease is very often diagnosed in the first years of life of the patient. While there is great clinical heterogeneity among patients with CGD, most develop bacterial infections particularly produced by catalase-positive microorganisms, and fungal diseases. Such infections are severe and recurrent, and often involve the formation of granulomas. The most common clinical manifestations are lymphadenitis, pneumonia and hepatosplenomegalia5,7.
The laboratory diagnosis of CGD is based on the demonstration of absence of phagocyte oxidizing activity. Many different diagnostic tests have been developed to this effect (direct measurement of superoxide production, ferrocytochrome reduction, the nitroblue tetrazolium test (NBT), and the oxidation test via flow cytometry)8-10. Flow cytometry is presently the most widely used technique, due to its greater objectiveness and reproducibility. The treatment options primarily aim to deal with the intercurrent infections and prevent the latter through antibiotic prophylaxis particularly involving drugs capable of penetrating phagocytic cells (trimethoprim-sulphamethoxazole)11 and antifungals (itraconazole)12. Gamma-interferon (IFN-γ) as a nonspecific stimulant of phagocytic cells is used by many centers for treating the more severe infections13-15. Hematopoietic cell progenitors transplants presently offer the only possibility of healing the disease, by replacing the patient immune response cells with normal cells16,19. However, MHC compatibility with the donor is required (the latter preferentially being a sibling unaffected by the disease), since the host T cell response is normal. Furthermore, the immune suppressive treatment required to avoid graft versus host disease (GVHD) increases the risk of infections. In future, gene therapy, involving replacement of the mutant gene with the normal gene, may prove to be the most effective treatment option20-22.
The present study describes the clinical characteristics, the infections detected, and the microorganisms involved in a group of pediatric patients diagnosed with CGD in the period 1980-2005.
MATERIAL AND METHODS
The study data were obtained by reviewing the clinical records of the patients diagnosed of chronic granulomatous disease (CGD) during childhood (age 0-16 years) in the period between January 1980 and December 2005, in Vall d'Hebron University Hospital in Barcelona (Spain). One of the patients was diagnosed and is subjected to control in Sabadell Hospital (Spain).
The study comprised a total of 13 patients, with collection of the following data at diagnosis: age, clinical manifestations, implicated microorganism (in the event of an infectious process), and methods used to confirm the suspected diagnosis.
In most cases, the diagnosis was confirmed using the flow cytometry oxidative test with dihydrorhodamine (9/13 cases, since 1995), while the first cases were diagnosed with the nitroblue tetrazolium test (NBT). Since the first description of the mutant genes implicated in CGD, familial genetic studies are made for diagnostic confirmation, the study of carriers (X-linked forms), and for genetic counseling in the context of prenatal diagnosis, where applicable. The mutational studies were made in the Department of Blood Cell Research in Amsterdam (Dr. D. Roos), in the Division of Immunology of the Kinderspital in Zurich (Dr. J.P. Hossle), and recently in the Immunology Laboratory of La Paz Hospital in Madrid (Spain) (Dr. A. Ferreira).
The flow cytometry oxidative test is based on detection of the fluorescence generated by dihydrorhodamine 123 (DHR) upon activation by the NADPH oxidative system of the previously activated phagocytic cells (or non-activated cells, used as negative controls). Such fluorescence can be detected and measured by the flow cytometer, and is compared between the non-stimulated cells and the stimulated cells yielding an index for measuring the production of these metabolites8.
Regarding the treatment provided, we documented the use of prophylactic antibiotic treatment with cotrimoxazole and/or itraconazole, or the administration at some time of gamma-interferon by subcutaneous route.
A review was also made of all the infectious processes experienced by the patients in the course of the disease, together with the microorganisms involved. Finally, the life course of CGD was examined in each case.
RESULTS
The sample characteristics are summarized in Table 1.
Data were collected on 13 patients (all males) diagnosed of CGD in childhood.
Median age at the time of diagnosis was three years (range 3-137 months). Forty-five percent of the cases were diagnosed before three years of age, and over 90 % before 6 years of age. In one case the diagnosis was established at 12 years of age.
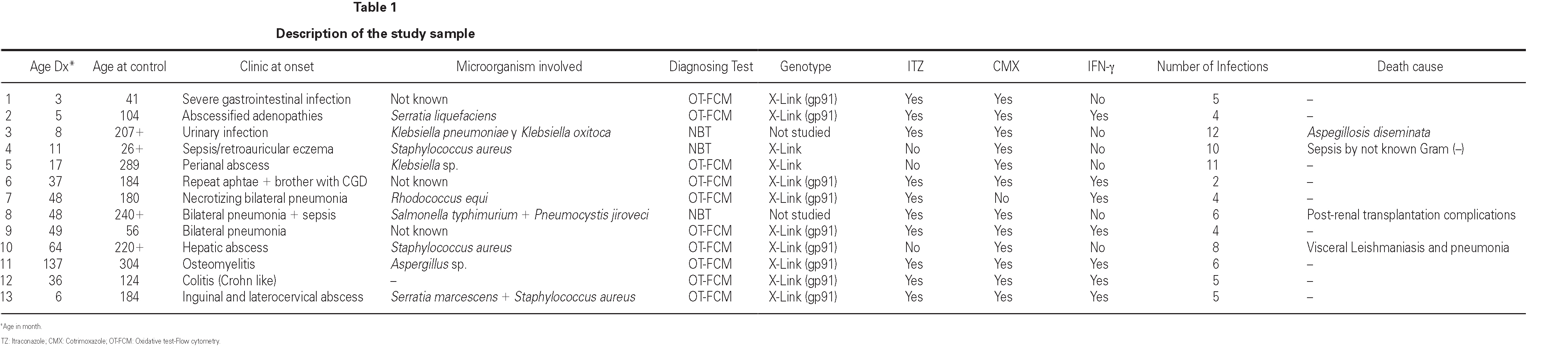
The cases diagnosed before 1995 (10/13) were initially identified by the nitroblue tetrazolium test (NBT). All but four of these cases, and those diagnosed posteriorly, were confirmed by means of the flow cytometry oxidative test (10/13) and using genetic techniques (gp91phox, X-linked transmission) in 9/13. A carrier assessment was made of the female relatives using flow cytometry (fig. 1), together with genetic study in those cases where the mutation was known. All mothers of the patients are heterozygous carriers of the mutation, and an additional four carriers have been identified in two of the families studied. One mother with the mutation (patient 9) presented lesions compatible with cutaneous lupus erythematosus.
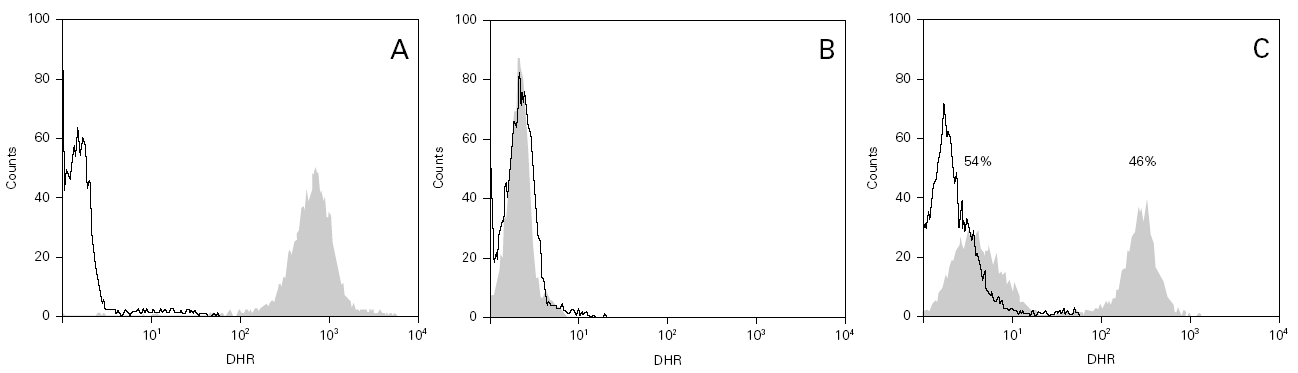
Figure 1.--Flow cytometry in patient 7: carriers sibling and healthy control. Grey zone: patient's neutrophyls. Black line: not activated neutrophyls of control people. A. Normal study. B. Absence of oxidative capacity (diagnose of GCD). C. Two population of neutrophyls with different oxidative capacity (carrier of X-link)
The clinical manifestations at the time of diagnosis were: abscesses and/or abscessified adenopathies in four cases, yielding as causal microorganisms S. aureus (n = 2), S. liquefaciens, S. marcescens and Klebsiella sp.; and pneumonia in three cases with the isolation in two of them of R. equi, and S. typhimurium (the latter as coinfection with P. jiroveci). The other patients initially manifested with different disorders such as osteomyelitis due to Aspergillus sp., urinary infection produced by Klebsiella sp., sepsis due to S. aureus, Crohn-like inflammatory bowel disease, severe acute gastroenteritis, and oral aphthae, without isolation of the causal microorganisms in these last cases.
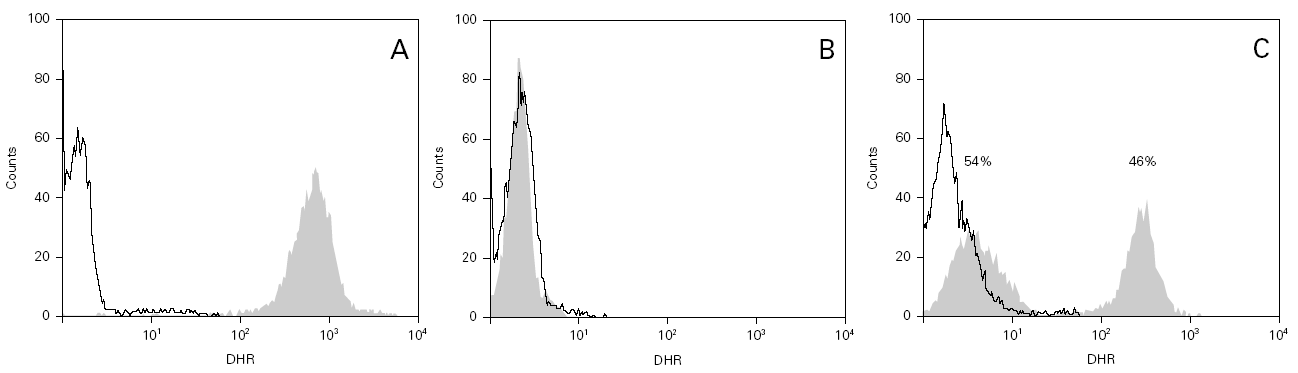
During the course of follow-up, the patients suffered a total of 88 infectious processes (0.27 episodes per patient and year): abscesses (n = 26), lymphadenitis (n = 12), pneumoniae (n = 10), gastroenteritis (n = 7), sepsis (n = 6), osteomyelitis, acute otitis, urinary infection and muguet (n = 3 every one), tonsillitis, typhoid fever and fever syndrome (n = 2 every one) and bacterial conjunctivitis, cryptytis, orchitis, pustulosis and infected subcutaneous granuloms (n = 1 every one) (table 1) (fig. 2).
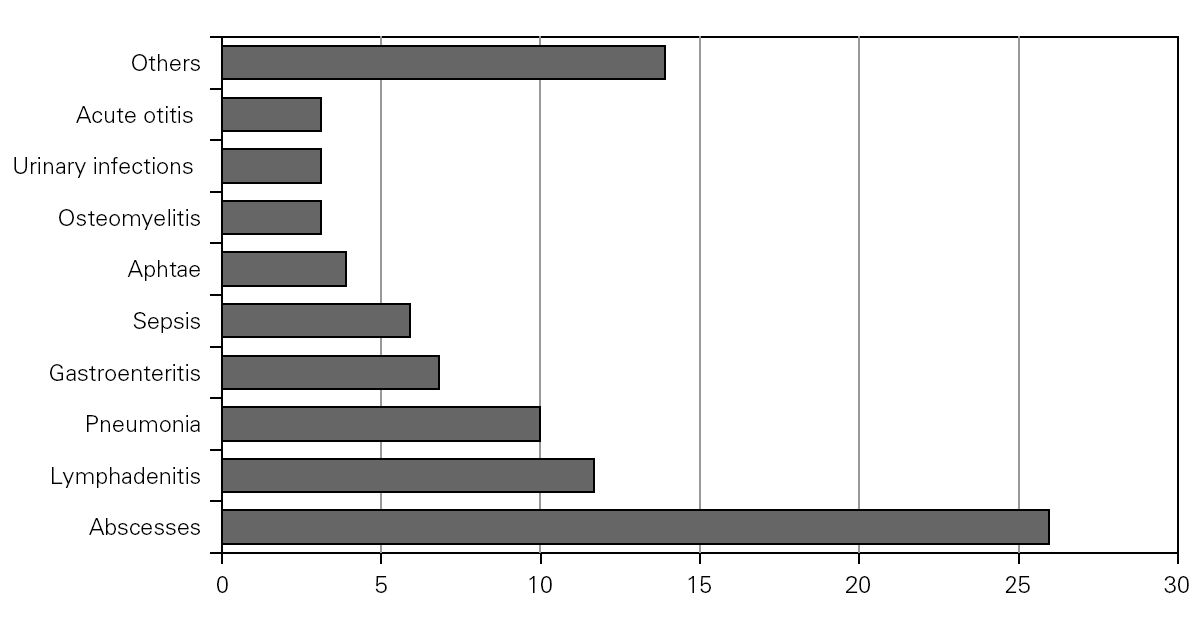
Figure 2.--Infectious processes documented since diagnosis in the patients studied.
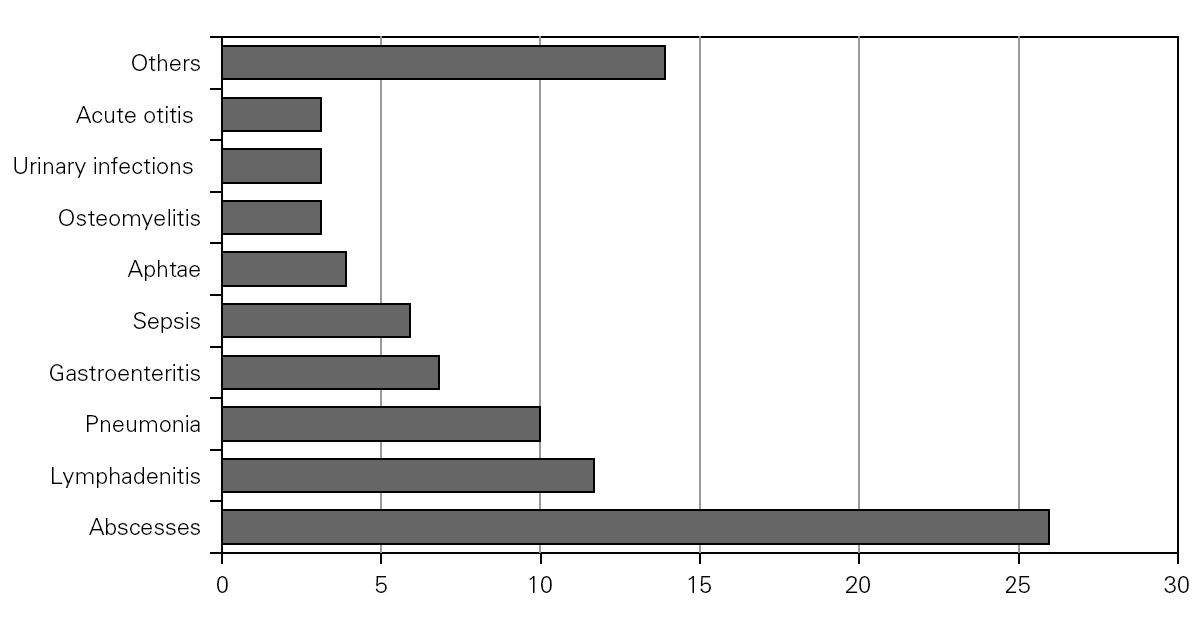
Causal microorganisms were isolated in 43 of these 88 infectious episodes (49 %), with a total of 49 isolates: Aspergillus sp. (n = 10), Staphylococcus sp. (n = 7), Salmonella sp. (n = 6), Serratia sp. (n = 5) Pseudomonas aeruginosa (n = 4), Klebsiella sp. (n = 4), Proteus sp. (n = 3), Leishmania sp. (n = 2) and others (n = 8) (fig. 3).
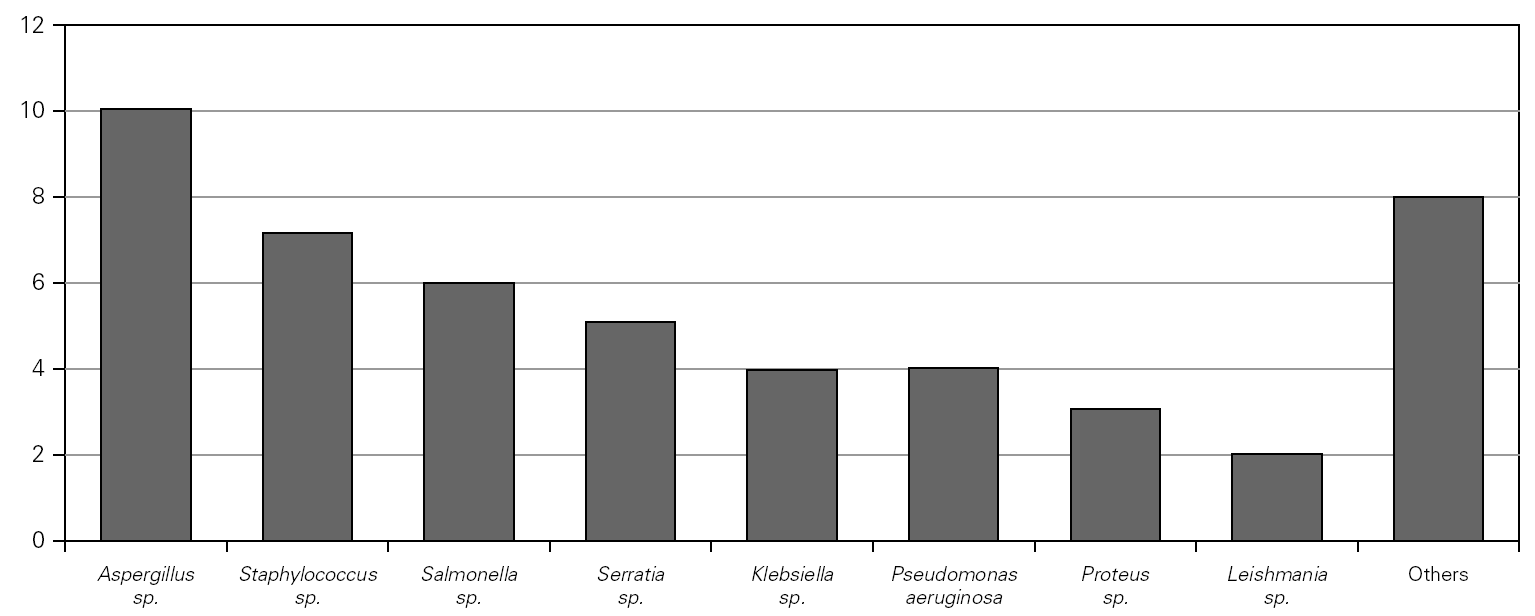
Figure 3.--Microorganisms isolated in the infectious processes cited in figure 2.
In relation to antibiotic prophylaxis, three patients received only cotrimoxazole (CTMX), while 10 received both antibiotics. Seven patients received IFN-γ at some time during the course of the disease; all of them also received antibiotic prophylaxis with CTMX and itraconazole.
At the end of the study, four patients had died: three due to infection (1 case of sepsis due to an unidentified gramnegative bacillus, one case of disseminated aspergillosis, and one case of visceral leishmaniasis with associated hemophagocytic syndrome), and one due to post-kidney transplant complications. The median patient age at the time of death was 18 years (range 26-240 months).
DISCUSSION
Chronic granulomatous disease (CGD) is a primary immune deficiency resulting from a genetic defect in phagocyte oxidative activity, and gives rise to slow-evolving granulomas and encapsulated infections. Due to the low prevalence of CGD in the general population and the variable severity of the clinical manifestations, the diagnosis of the disease is sometimes established late a fact that can lead to sequelae that prove difficult to resolve (fig. 4). Diagnostic suspicion in the case of a child with the described clinical manifestations (frequent pneumonia, slowly resolving skin abscesses, adenitis with spontaneous fistulization or inflammatory bowel disease with granulomas) and usually presenting leukocytosis and hyper-gammaglobulinemia, should lead to perform the adequate studies to either discard or confirm the diagnosis of CGD. In this case, antibiotic prophylaxis and the adoption of hygiene-sanitary measures to avoid new infections are essential considerations in order to modify the patient prognosis.
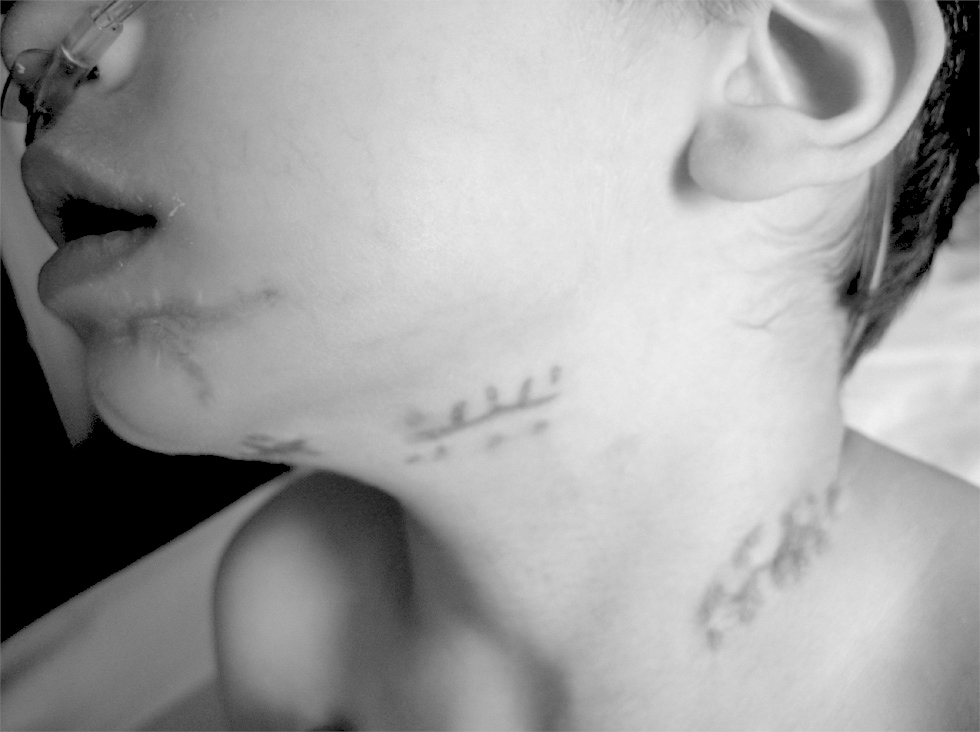
Figure 4.--Scarring corresponding to abscessified adenitis in patient 9.
CGD is generally diagnosed in childhood, since the X-linked form of the disease (XL) is the most common and also the most serious presentation of the disorder. Some patients mainly those presenting an autosomal recessive (AR) form of the disease can be diagnosed in adult life23. In our series, almost one-half of the patients were diagnosed before three years of age, and over 90 % of all cases were identified before 6 years of age. Only one patient was identified at 12 years of age, though in this case there were also compatible clinical manifestations of infection since early infancy. The above may be explained by the fact that all of our subjects presented an XL form of the disease in coincidence with larger reviews made in other geographical settings3.
Although the nitroblue tetrazolium test (NBT) is the most readily available diagnostic method, since 1994-1995 the standard technique has been flow cytometry oxidative test. The use of flow cytometry to determine phagocyte oxidative capacity allows more sensitive and reproducible detection of affected patients and carriers, and can indicate the nature of the underlying molecular defect24. In our series, flow cytometry oxidative test was used in 10 of the 13 patients. The other three patients were studied before 1995, which is the year in which the flow cytometric method was introduced in our center, and posterior oxidative test proved impossible for different reasons.
The absence of phagocytic oxidative capacity in patients with CGD makes them vulnerable to bacterial and fungal infections in different locations. In any case, the increased susceptibility to infection is quite limited to a concrete range of microorganisms a fact that should contribute to suspect the diagnosis3. In our series, the most common clinical manifestations at the time of diagnosis were abscessified granulomata in soft tissues (4/13 cases) and lung involvement in the form of pneumonia (3/13 cases). These results coincide with those of other larger reviews in which pneumonia and adenitis, and skin abscesses, were found to be the main manifestations at diagnosis. Regarding the implicated microorganisms, our observations likewise coincide with the descriptions in the literature, with a predominant presence of S. aureus, S. typhimurium, Serratia sp. and Aspergillus sp. This distribution of infections and implicated microorganisms persists throughout the evolution of the disease, with a tendency over time to increase the incidence of cutaneous abscesses and the implication of Aspergillus sp. The data obtained in our series coincide with those of larger reviews, where pneumonias were observed in 80 % of the patients, and adenitis and skin abscesses in 50 %. In turn, Aspergillus sp. and S. aureus were the most common causal microorganisms3.
It should be noted that one mother (a heterozygous carrier of the mutation) presented lesions compatible with cutaneous lupus erythematosus a circumstance that has been reported before in the reviewed literature25,26.
The usefulness of trimethoprim-sulphamethoxazole (CTMX) as antimicrobial prophylaxis in patients with CGD has been well established since the early 1990s, when a significant reduction was achieved in bacterial infections in these patients (both XL and AR forms)11. All of our patients received such treatment from the time of diagnosis. However, the demonstration that preventive treatment with CTMX does not modify the incidence of fungal processes led to the evaluation of itraconazole as prevention against Aspergillus sp. disease. In 2003, Gallin et al.12 conducted a double blind study comparing itraconazole versus placebo, in which the former was seen to reduce the incidence of Aspergillus sp. in pediatric patients with CGD12. Nevertheless, it must be stressed that no statistically significant differences were observed (possibly due to the limited number of patients involved), and that monitorization of the potential side effects (mainly skin rash and transaminase elevations) is required. In our series, 10/13 patients received prophylactic treatment with this antifungal drug, resulting in no side effects requiring treatment suspension.
The effectiveness of IFN-γ is subject to debate, since the exact mechanism by which it acts is not known, and the subcutaneous route of administration involved makes it more difficult to use in children. However, any mechanism capable of boosting activation of some of the other bactericidal mechanisms of phagocytic cells may be of help in eliminating intracellular infections the latter being the most frequent processes in CGD. Studies in animals have been unable to demonstrate that IFN-γ action takes place through an increase in the oxidative capacity of macrophages27,28. However, there appears to be an increased production of mRNA encoding for p47phox though it is not clear whether this is able to account for the observed reduction in infectious processes in these patients 29. In turn, a number of studies have suggested clinical benefit with this drug in the case of serious infections in patients with CGD30-31. We start therapy with IFN-γ in all patients, together with antibacterial and antifungal prophylactic measures, at the time of diagnosis. Posteriorly, and in the event of a good clinical course, such medication is suspended and reintroduced in cases presenting severe intracellular infections (Aspergillus sp., R. equi, etc).
Global mortality in our study was close to 30 %, which is higher than the figure reported in the largest series published to date3. In any case, the fact that all of our patients presented X-linked forms of the disease could constitute a source of bias in this sense, since mortality appears to be greater and occurs earlier in this subgroup of patients3.
In conclusion, it should be stressed that high clinical suspicion and flow cytometry are the keys to the diagnosis of CGD and detection of carrier relatives. In turn, it is important to establish specific antibacterial and antifungal prophylaxis, together with medical controls and the adoption of hygiene-sanitary measures to prevent severe infections. Lastly, while IFN-γ has been used intermittently in our series, its effectiveness in patients of this kind remains controversial.
Correspondence:
Dr. T. Español-Boren
Unidad de Inmunología
Hospital Universitario Vall d'Hebron
Passeig de la Vall d'Hebron 119-129
08035 Barcelona (Spain)
Phone: + 34-932746832
Fax: + 34-932746831
E-mail: tespanol@vhebron.net
