



Common variable immunodeficiency (CVID) is characterised by hypogammaglobulinaemia and a broad clinical spectrum, mainly showing recurrent bacterial infections accompanied sometimes by increased susceptibility to chronic lung disease, autoimmunity, and neoplastic diseases.
ObjectivesTo evaluate the clinical and immunological characteristics of patients with CVID in Mexico.
MethodsThis is a retrospective analysis of 43 patients with CVID from the Immunology Division of seven different reference centres in Mexico. Patients were diagnosed according to the diagnostic criteria of the European Society for Immunodeficiency Diseases. We collected demographics, clinical and immunological data from each patient and a statistical analysis was performed.
ResultsThere were 23 (53.5%) male and 20 (46.5%) female patients. Median age at onset of disease was 13.7 years, and median age at diagnosis was 19 years. Average delay in diagnosis was 12.5 years. The median total serum levels of IgG, IgM, and IgA at diagnosis were 175, 18, and 17.8mg/dL, respectively. The mean percentage of CD19+ B cells was 8.15%. Sinusitis (83%), pneumonia (83%), gastrointestinal infection (70%), and acute otitis media (49%) were the most common manifestations. Bronchiectasis was present in 51% of the patients, 44% manifested non-infectious chronic diarrhoea, and 70% experienced weight loss. Autoimmunity was present in 23% of the patients; haemolytic anaemia and autoimmune thrombocytopenic purpura were the most common presentations. Allergy was present in 30.2% of patients, with allergic rhinitis and asthma being the most frequent types. Two patients developed malignancy. All the patients received Intravenous immunoglobulin (IVIG) as a fundamental part of the treatment at a mean dose of 408mg/kg.
ConclusionThis is the first cohort of CVID reported in Mexico We found that infection diseases were the most frequent presentations at onset. Moreover, patients had an average diagnosis delay of twelve years and thus a major prevalence of bronchiectasis. We suggest performing an extended analysis of patients with CVID patients in other Latin American countries.
Common variable immunodeficiency (CVID) is a primary immunodeficiency syndrome characterised by hypogammaglobulinaemia and recurrent bacterial infections.1 It represents the most frequent symptomatic primary immunodeficiency in North America and Europe.2 Its diagnosis requires a history of recurrent or chronic bacterial infections, a significant reduction of immunoglobulin G (IgG) (>2 standard deviation), a reduction of immunoglobulin A (IgA) or immunoglobulin M (IgM), normal or low counts of B cells, as well as autoimmune, lymphoproliferative and/or granulomatous disease. Other causes of hypogammaglobulinaemia should be excluded as indicators of CVID.3,4
The clinical spectrum of CVID is quite broad, and it may onset at any time in life.5 In CVID patients, increased susceptibility to chronic lung disease and autoimmune, gastrointestinal, neoplastic, and inflammatory disorders have been reported. Bronchiectasis is a particularly common medical problem, leading to recurrent hospitalisations and severe respiratory symptoms.6Streptococcus pneumoniae and Haemophilus influenzae are the most common bacteria affecting the respiratory tract during CVID. Giardia, Salmonella and Campylobacter were commonly isolated from patients with diarrhoea.7 Standard treatment for CVID requires periodic administration of intravenous immunoglobulin (IVIG). The aim of this study was to describe the clinical and immunological features of CVID in a group of patients in Mexico.
Patients and methodsPatientsThe records of 52 active CVID patients from the Immunology Division of seven different reference centres in Mexico were studied. CVID was diagnosed according to the diagnostic criteria of the European Society for Immunodeficiency Diseases (ESID). Information from each CVID subject was collected by means of a structured questionnaire applied by a single physician: demographics, age at onset of symptoms, age at diagnosis, pedigree, clinical manifestations, current infections, autoimmunity, lymphoproliferative disease, allergy and malignancies, lymphocyte subsets and immunoglobulin levels at the first clinical visit, and route and dosage of immunoglobulin.
The protocol was reviewed and approved by the appropriate local Ethics and Research Committees in accordance with the guidelines of the International Conference on Harmonization Good Clinical Practice and the Declaration of Helsinki, to access details of clinical records, and on the publication of patient data on their manuscript for research and divulgation purposes for the scientific community.
Statistical analysisResults are shown as mean±sd (if the variable is normally distributed) and median (interquartile range) for continuous variables. Dichotomous and nominal variables were expressed as frequencies and percentages. All two-sided P-values <0.05 were considered to be statistically significant. Data were analysed using the Statistical Package for Social Science (SPSS) for Windows (version 12.00, SPSS Inc., Chicago, IL, USA).
ResultsThe data from the 52 patients from 7 centres were reviewed in order to ascertain that each individual met the diagnostic criteria for CVID. A total of nine patients were removed for the following reasons: hypogammaglobulinaemia secondary to thymectomy (2), X-linked lymphoproliferative syndrome (2), transient hypogammaglobulinaemia of infancy (2), recessive agammaglobulinaemia (1), ataxia telangiectasia (1), and X-linked agammaglobulinaemia (1).
DemographicsDiagnosis was confirmed for 43 patients; 23 (53.5%) were males, and 20 (46.5%) were females; two patients were sisters. The median age at the onset of disease was 13.7 years (9.37–18.0), and the median age at diagnosis was 19 years (14.5–23.5), thus indicating that the average delay in diagnosis was 12.5 (10.7–26.8) years. The mean follow-up time was 24.5 years (±15) (Table 1).
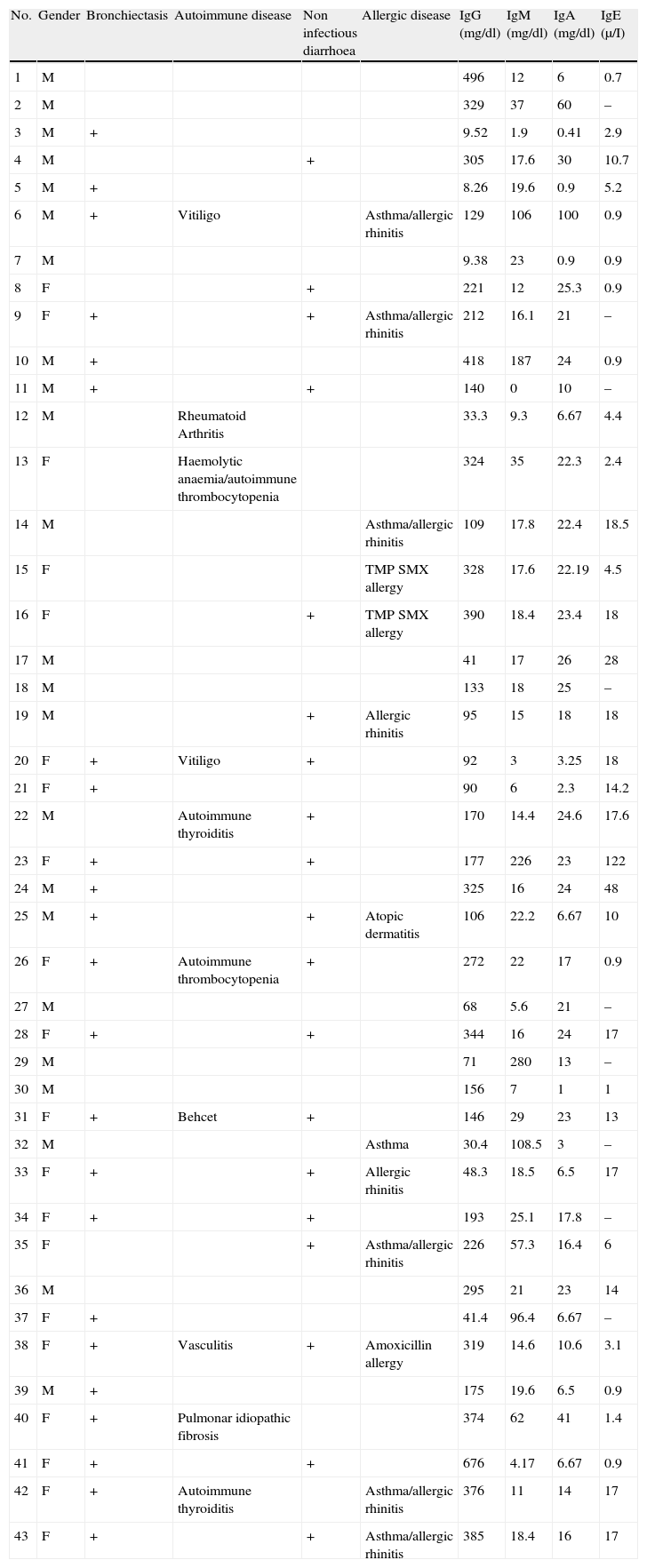
Clinical and laboratory data of the cohort.
| No. | Gender | Bronchiectasis | Autoimmune disease | Non infectious diarrhoea | Allergic disease | IgG (mg/dl) | IgM (mg/dl) | IgA (mg/dl) | IgE (μ/I) |
| 1 | M | 496 | 12 | 6 | 0.7 | ||||
| 2 | M | 329 | 37 | 60 | – | ||||
| 3 | M | + | 9.52 | 1.9 | 0.41 | 2.9 | |||
| 4 | M | + | 305 | 17.6 | 30 | 10.7 | |||
| 5 | M | + | 8.26 | 19.6 | 0.9 | 5.2 | |||
| 6 | M | + | Vitiligo | Asthma/allergic rhinitis | 129 | 106 | 100 | 0.9 | |
| 7 | M | 9.38 | 23 | 0.9 | 0.9 | ||||
| 8 | F | + | 221 | 12 | 25.3 | 0.9 | |||
| 9 | F | + | + | Asthma/allergic rhinitis | 212 | 16.1 | 21 | – | |
| 10 | M | + | 418 | 187 | 24 | 0.9 | |||
| 11 | M | + | + | 140 | 0 | 10 | – | ||
| 12 | M | Rheumatoid Arthritis | 33.3 | 9.3 | 6.67 | 4.4 | |||
| 13 | F | Haemolytic anaemia/autoimmune thrombocytopenia | 324 | 35 | 22.3 | 2.4 | |||
| 14 | M | Asthma/allergic rhinitis | 109 | 17.8 | 22.4 | 18.5 | |||
| 15 | F | TMP SMX allergy | 328 | 17.6 | 22.19 | 4.5 | |||
| 16 | F | + | TMP SMX allergy | 390 | 18.4 | 23.4 | 18 | ||
| 17 | M | 41 | 17 | 26 | 28 | ||||
| 18 | M | 133 | 18 | 25 | – | ||||
| 19 | M | + | Allergic rhinitis | 95 | 15 | 18 | 18 | ||
| 20 | F | + | Vitiligo | + | 92 | 3 | 3.25 | 18 | |
| 21 | F | + | 90 | 6 | 2.3 | 14.2 | |||
| 22 | M | Autoimmune thyroiditis | + | 170 | 14.4 | 24.6 | 17.6 | ||
| 23 | F | + | + | 177 | 226 | 23 | 122 | ||
| 24 | M | + | 325 | 16 | 24 | 48 | |||
| 25 | M | + | + | Atopic dermatitis | 106 | 22.2 | 6.67 | 10 | |
| 26 | F | + | Autoimmune thrombocytopenia | + | 272 | 22 | 17 | 0.9 | |
| 27 | M | 68 | 5.6 | 21 | – | ||||
| 28 | F | + | + | 344 | 16 | 24 | 17 | ||
| 29 | M | 71 | 280 | 13 | – | ||||
| 30 | M | 156 | 7 | 1 | 1 | ||||
| 31 | F | + | Behcet | + | 146 | 29 | 23 | 13 | |
| 32 | M | Asthma | 30.4 | 108.5 | 3 | – | |||
| 33 | F | + | + | Allergic rhinitis | 48.3 | 18.5 | 6.5 | 17 | |
| 34 | F | + | + | 193 | 25.1 | 17.8 | – | ||
| 35 | F | + | Asthma/allergic rhinitis | 226 | 57.3 | 16.4 | 6 | ||
| 36 | M | 295 | 21 | 23 | 14 | ||||
| 37 | F | + | 41.4 | 96.4 | 6.67 | – | |||
| 38 | F | + | Vasculitis | + | Amoxicillin allergy | 319 | 14.6 | 10.6 | 3.1 |
| 39 | M | + | 175 | 19.6 | 6.5 | 0.9 | |||
| 40 | F | + | Pulmonar idiopathic fibrosis | 374 | 62 | 41 | 1.4 | ||
| 41 | F | + | + | 676 | 4.17 | 6.67 | 0.9 | ||
| 42 | F | + | Autoimmune thyroiditis | Asthma/allergic rhinitis | 376 | 11 | 14 | 17 | |
| 43 | F | + | + | Asthma/allergic rhinitis | 385 | 18.4 | 16 | 17 |
The median total serum levels of IgG, IgM, and IgA at diagnosis were 175 (160–253), 18.0 (20.8–57.5), and 17.8 (13.1–23.8)mg/dL, respectively (Table 1). Lymphocyte subset distribution was also assessed. The median expression for CD3 was 77% (67–80), 34% (29–45) for CD4, and 37% (35–47) for CD8. The median CD19+ B cells was 8.15% (6.7–13.2).
InfectionsInfections were the most common clinical manifestation. All the patients had recurrent infections as the initial manifestation, except for two patients having autoimmunity. Among infections, the four most frequent were sinusitis (36, 83%), pneumonia (36, 83%), gastrointestinal infection (30, 70%), and acute otitis media (21, 49%). The less frequent were urinary tract infections (7, 16%), meningitis (6, 14%), mastoiditis (3, 7%), and vulvovaginitis (3, 7%). Culture-proven sepsis, stomatitis, osteomyelitis, cellulitis, and perianal abscess were each found in two patients (5%). Septic arthritis, brain abscess, mucocutaneous herpes simplex virus, peritonitis, fasciitis, and endocarditis were each found in one patient (2%).
Organisms most commonly isolated from the respiratory tract were Streptococcus pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus viridans. Giardia lamblia and Salmonella enterica were the most frequent causes of gastrointestinal infections. The remaining infectious agents were diverse and included gram-negative bacteria, mycobacteria, and fungus (Table 2).
Aetiological agents of infectious diseases.
| Etiologic Agent | Pneumonia | Diarrhoea | Sinusitis | Meningitis | Otitis media | Mastoiditis | Total |
| Giardia lamblia | 7 | 7 | |||||
| Streptococcus pneumoniae | 6 | 6 | |||||
| Pseudomonas aeruginosa | 4 | 1 | 1 | 6 | |||
| Staphylococcus aureus | 4 | 1 | 5 | ||||
| Streptococcus viridans | 3 | 1 | 4 | ||||
| Burkholderia cepacia | 3 | 3 | |||||
| Haemophilus influenzae | 2 | 1 | 3 | ||||
| Streptococcus mitis | 2 | 1 | 3 | ||||
| Streptococcus salivarius | 1 | 2 | 3 | ||||
| Salmonella enterica gp B | 3 | 3 | |||||
| Candida albicans | 2 | 2 | |||||
| Escherichia coli | 2 | 2 | |||||
| Streptococcus milleri | 1 | 1 | |||||
| Klebsiella oxytoca | 1 | 1 | |||||
| Klebsiella pneumoniae | 1 | 1 | |||||
| Serratia marcescens | 1 | 1 | |||||
| Enterobacter cloacae | 1 | 1 | |||||
| Mycobacterium tuberculosis | 1 | 1 | |||||
| Comamonas testosteroni | 1 | 1 | |||||
| Morganella morganii | 1 | 1 | |||||
| Pneumocystis jiroveci | 1 | 1 | |||||
| Coccidioides immitis | 1 | 1 | |||||
| Neisseria sp. | 1 | 1 | |||||
| Corynebacterium sp. | 1 | 1 | |||||
| Herpes virus | 1 | 1 | |||||
| Enterococcus faecium | 1 | 1 | |||||
| Citrobacter freundii | 1 | 1 | |||||
| Proteus mirabilis | 1 | 1 | |||||
| Chysosporium sp | 1 | 1 | |||||
| Criptosporidium sp | 1 | 1 | |||||
| Entamoeba coli | 1 | 1 | |||||
| Campylobacter jejuni | 1 | 1 | |||||
| Total/% | 37/(55.2%) | 19/(28.3%) | 7 (10.4%) | 2/(2.8%) | 1/(1.4%) | 1/(1.4%) | 67/(100%) |
Note: Only infectious episodes in which an aetiological agent was identified are shown.
Clinical and serological viral infections were positive for Epstein Barr virus (n=5), varicella-zoster virus (n=3), and cytomegalovirus (n=1).
Ninety-five percent of the patients were hospitalised at least once due to infection. The median number of hospitalisations per patient was 3.1 (3.0–7.14), and the overall median length of hospitalisation was 50 days (37.3–80.9) per patient.
Pulmonary diseaseBronchiectasis, detected through high-resolution computed tomography, was found in 22 (51%) patients at the time of evaluation for this study. Of the patients who suffered from pneumonia, 61% developed bronchiectasis (Table 1).
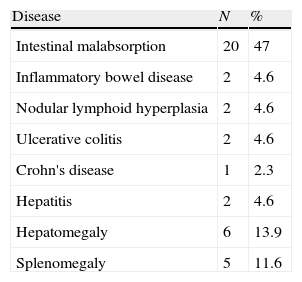
Gastrointestinal diseaseNineteen (44%) patients showed non-infectious chronic diarrhoea, and 15 out of 19 (79%) patients experienced weight loss. Biopsies performed in two patients with chronic diarrhoea revealed eosinophilic infiltration in one patient, and eosinophilic infiltration and lymphoid hyperplasia in the other (Tables 1 and 3).
AutoimmunityTwo patients showed autoimmunity as first manifestation; both suffered from autoimmune thrombocytopenic purpura (ATP), and one additionally suffered from autoimmune haemolytic anaemia.
A family history of autoimmunity was present in three patients: systemic lupus erythematous in one, and idiopathic thrombocytopenic purpura in two. Autoimmunity occurred in 10 (23.2%) patients, including seven females and three males. This finding was evident from eight different diseases: haemolytic anaemia (1), vitiligo (2), thyroiditis (2), arthritis (1), vasculitis (1), Behcet's disease (1), and idiopathic pulmonary fibrosis (1). One patient had haemolytic anaemia and autoimmune thrombocytopenic purpura. The median age at autoimmune disease onset was 12.5 (10.7–26.7) years, and the median age at diagnosis was 14 (11.2–27). Two out of four patients with splenomegaly had autoimmune cytopenia (Table 1).
AllergyAllergy was present in 13 (30.2%) patients. Manifestations were allergic rhinitis and asthma (6), rhinitis (2), atopic dermatitis (1), asthma and atopic dermatitis (1), and drug allergy (3). Median IgE level was 8 (5.8–20.9)μI/mL (Table 1).
Neoplastic diseaseOne patient suffered from myeloid metaplasia with liver and spleen infiltration, and cutaneous T-cell lymphoma.
Physical examinationTonsils were present in 34 (79.1%) patients, absent in three patients, and undocumented in two patients. Lymph nodes, liver, and spleen had increased size in 29, 4, and 4 patients, respectively.
TreatmentAll the patients started with intravenous immunoglobulin (IVIG) at diagnosis, and the mean dose was 408mg/kg (±130), and received prophylactic antibiotic with azithromycin.
DiscussionCommon variable immunodeficiency may onset at any time in life, both in childhood and old age.5 The delay in diagnosis in patients from Mexico was similar to that reported in other studies around the world.1,6,8,9
We report that CVID equally affects females and males, as previously described in other studies. The complexity of CVID has been evidenced by the wide spectrum of clinical presentations, and a genetic origin has been demonstrated in several cases. Our results may suggest a genetic background given that two sisters had CVID.
Serum immunoglobulin levels reported in the present study are similar to those reported previously.1 In our patients, we found an inverse relationship between CD4+ and CD8+ T cells, which is explained by the decreased number of CD4 T cells. This finding has been previously described in patients with CVID, and the reduction in CD4+ T cells has been associated with heterogeneous clinical features.11 No patient showed CD19+ percentages lower than 1%, as reported previously.12
Infection was the presenting manifestation in most of the cases in this study. The most frequent infections were present in the upper and lower respiratory tract, as reported previously.4,10,13–15 However, we found that all the organs and systems were affected. We also found a higher percentage of gastrointestinal infections among patients than other studies. Nevertheless, the aetiological agent was rarely isolated, and the diagnosis was predominantly clinical. Therefore, we cannot rule out intestinal autoimmunity.
Aetiological agents were diverse, most frequently identified agents were gram-positive bacteria from the respiratory tract, and Giardia lamblia from the gastrointestinal tract.1,12
Almost all the patients had been hospitalised due to infections, although we did not distinguish between hospitalisations before and after IVIG. As reported previously, however, the frequency of hospitalisations decreased after the IVIG treatment.16
The prevalence of bronchiectasis reported in this study was up to 20% higher than that reported by others.4,12,13 This difference may be explained by a longer delay in diagnosis. The development of pneumonia was associated with the presence of bronchiectasis, as described previously.4 Other authors have associated the presence of bronchiectasis to other factors, including the delay in diagnosis, a smaller number of B lymphocytes, and increased use of antibiotics or absence of replacement therapy with immunoglobulin. Although we did not perform an analysis of mortality, survival or quality of life, we would expect worse results for the three variables in our cohort in comparison to those reported without bronchiectasis.13
We found a higher prevalence of patients with chronic non-infectious diarrhoea as compared to previously reported studies.10,13,15,17 However, we did not have histopathology or cultures for all of the patients in the cohort. It is important to distinguish between infectious and autoimmune etiologic; the difference between them is subtle as they can both lead to chronic, even severe, diarrhoea characterised by weight loss, steatorrhoea, and malabsorption.18
The presence of autoimmunity in CVID patients was similar to that reported previously.15,18 Haemolytic anaemia (HA) and thrombocytopenic purpura (PTI) were the most prevalent autoimmune diseases; however, we also identified other autoimmune diseases, including neutropenia, arthritis, vitiligo, thyroiditis, vasculitis, and idiopathic pulmonary fibrosis. Autoimmunity could be the first manifestation in CVID.19 In cases with PTI and AH, we suggest ruling out CVID.20 Finally, we also found a family history of autoimmunity in some patients.18
Our CVID cohort presents with comparable symptoms and disorders as previously reported. We consider that this study could be further complemented with patients from other countries who are members of the Latin American Society for Immunodeficiencies (LASID) in order to perform a more robust analysis of CVID outcome in Latin America.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that they have followed the protocols of their health center to access details of clinical records, and on the publication of patient data on their manuscript for research and divulgation purposes for the scientific community.
Right to privacy and informed consentBeing a retrospective study, there is no informed consent of patients referred in this study. However, it has been authorized by the Ethics and Research Committees of the National Institute of Pediatrics.
Conflict of interestThe authors declare that they have no conflict of interest.
