





INTRODUCCION
Las inmunodeficiencias primarias (IDP) son defectos congénitos de alguno de los mecanismos de la respuesta inmunológica, lo que predispone a presentar infecciones con mayor frecuencia, de mayor gravedad y generalmente, difíciles de curar. El diagnóstico de las IDP 1-3 se ve dificultado y retrasado por la difundida creencia de su rareza, totalmente injustificada con los datos que disponemos en la actualidad (Registro Español IDP) 4. El retraso diagnóstico, que puede llegar a ser de varios años, condiciona una peor evolución clínica de los pacientes 5,6. Por todo ello, es importante insistir en la divulgación del conocimiento de signos de alarma y formas de presentación clínica de las IDP, y a la vez, facilitar las direcciones de los centros en los que se pueden realizar estudios específicos, en aras a mejorar el manejo y el pronóstico de estos pacientes. El origen genético de la mayoría de ellas, perfectamente dilucidado en mas de 100 IDP, posibilita además el diagnóstico de portadores y prenatal 7.
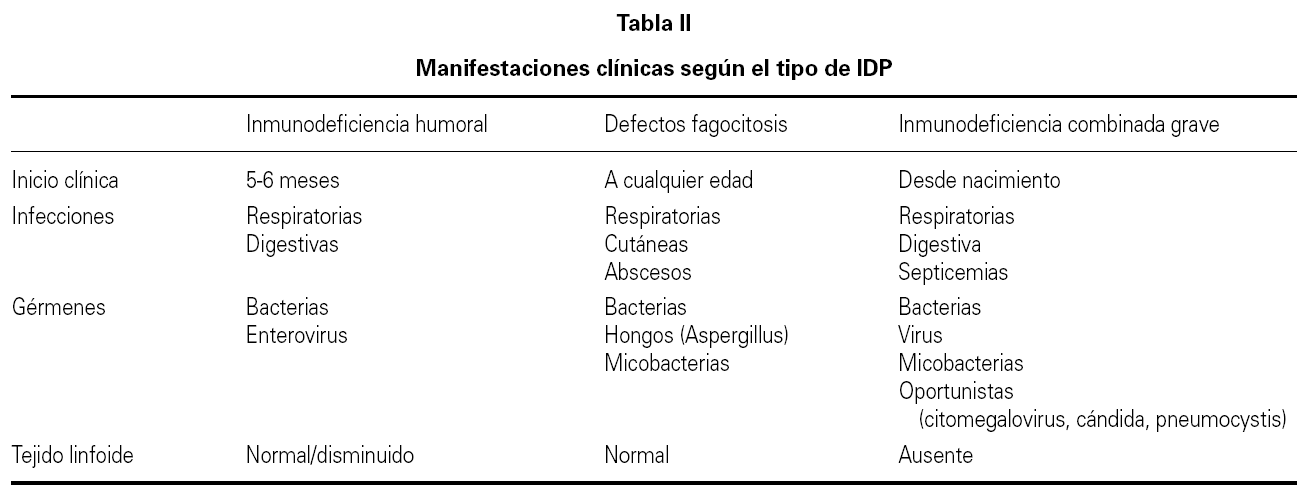
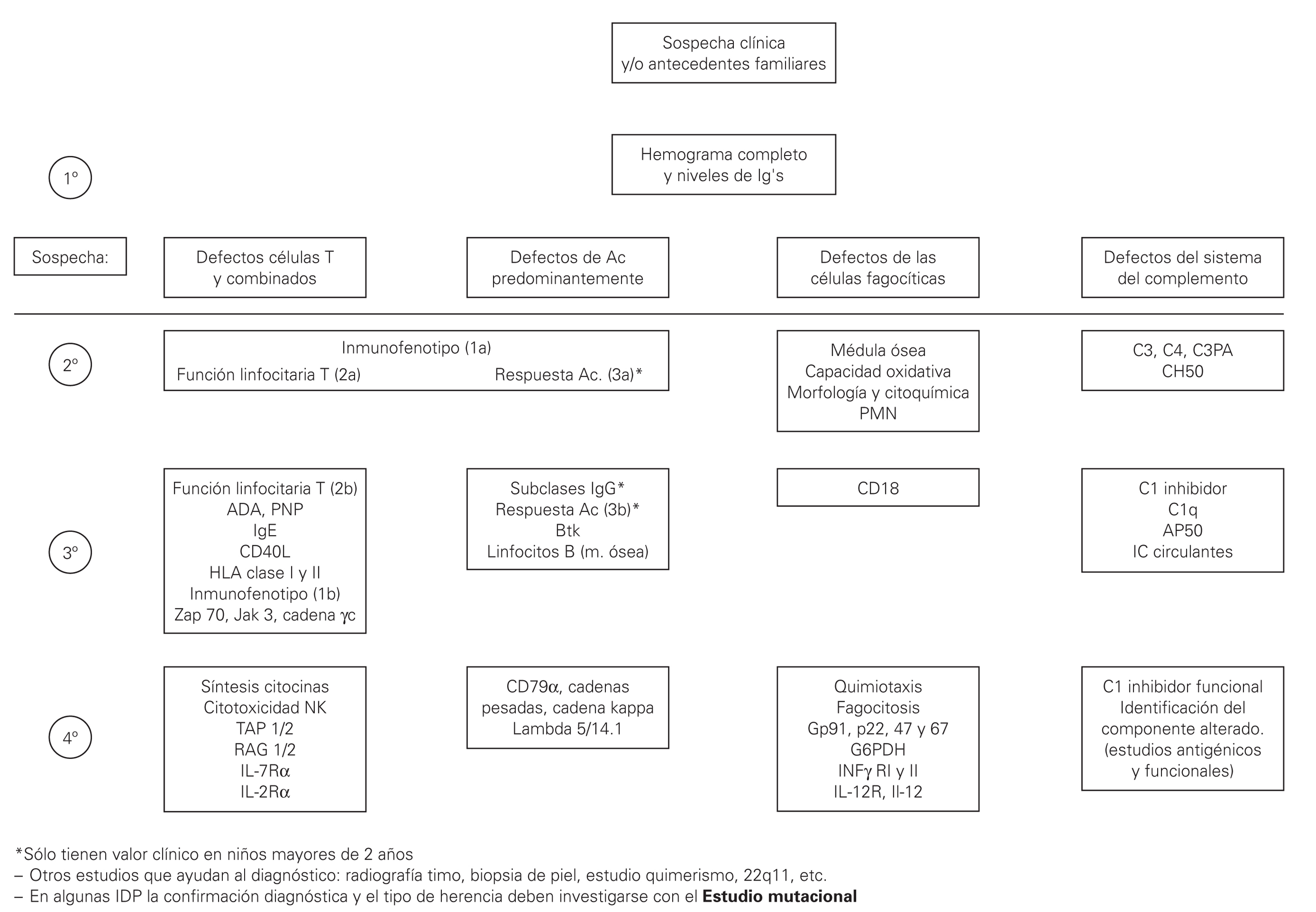
En el presente trabajo se incluyen unas tablas de orientación clínica general (tabla I), las manifestaciones clínicas según el defecto inmunológico (tabla II), los patrones clínicos con su diagnóstico diferencial (tabla III) y un algoritmo diagnóstico de las IDP (fig. 1). Se incluye además, un directorio de centros españoles con las pruebas diagnósticas que se realizan en cada uno de ellos.

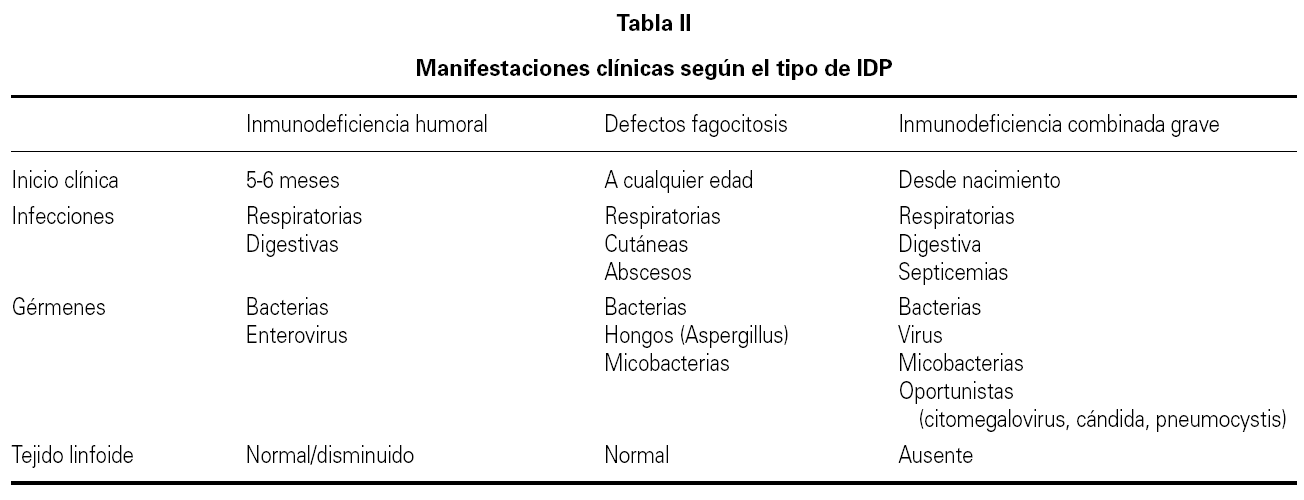

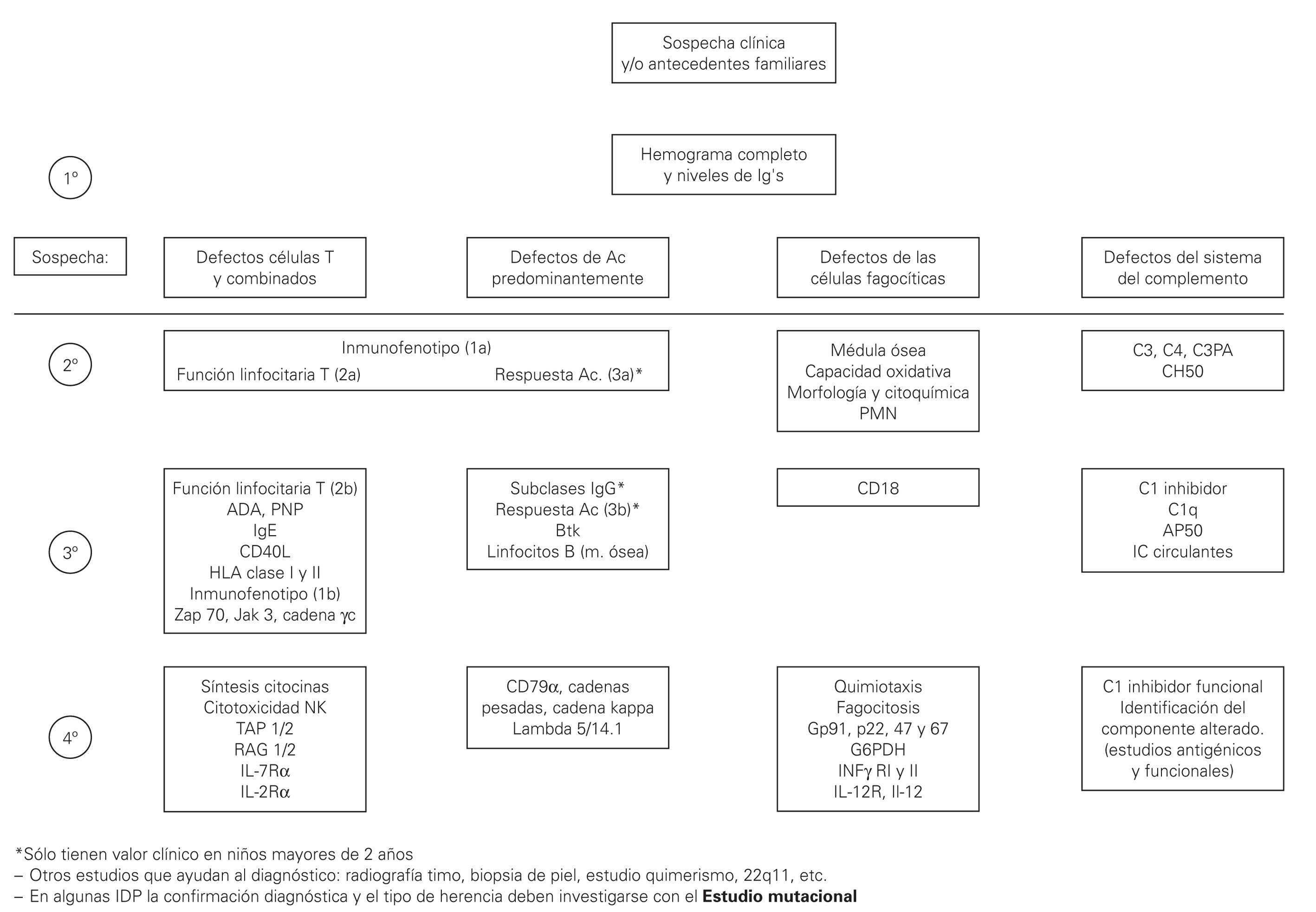
Figura 1.--Algoritmo diagnóstico de las IDP.
MATERIAL Y MÉTODOS
Con el objetivo de conocer los laboratorios donde se pueden realizar las determinaciones para obtener el diagnóstico de las IDP, se realizó una encuesta a los hospitales con laboratorios de Inmunología y a los miembros de las Unidades de Alergia e Inmunología clínica pediátrica.
RESULTADOS
Con las respuestas obtenidas podemos establecer que:
1. En nuestro país se pueden realizar los diagnósticos inmunológicos, y la mayoría de los estudios genéticos, de las IDP descritas hasta actualidad.
Sin embargo en primer lugar se debe tener la sospecha de que se trate de una de estas enfermedades y en segundo lugar hay orientar el estudio. No se pueden realizar todos los estudios en todos los pacientes con sospecha de IDP, por lo tanto es importante conocer las manifestaciones características de distintas formas de IDP (ver tablas) y seguir un algoritmo de estudio 9 (tabla IV), desde las determinaciones más frecuentes a los estudios genéticos específicos.

2. En todos los hospitales de 2.º y 3.er nivel se realizan: los niveles séricos de Inmunoglobulinas, la respuesta de anticuerpos (Ac) frente a las vacunaciones habituales en niños, niveles de algunas proteínas del complemento y la actividad hemolítica (CH50) y la valoración de las subpoblaciones linfocitarias.
En muchos casos una alteración significativa de estos parámetros, sin causa conocida, y siempre en relación a los valores normales para la edad 10-13, debe ya hacer sospechar una IDP y dar paso a un estudio mas específico, según el tipo de infecciones y patrón clínico de presentación.
3. En los centros hospitalarios de 3.er nivel se realiza la valoración de las subclases de la IgG; en algunos de ellos: La Paz y Gregorio Marañon (Madrid), Vall d'Hebron (Barcelona), Virgen del Rocío (Sevilla), Son Dureta (Palma de Mallorca) se detecta la respuesta de Ac frente a vacunas (Neumococo y Haemophilus), también en adultos. También se detectan moléculas de activación linfocitaria, receptores de algunas citocinas, etc.
4. El estudio de la función linfocitaria (respuesta a mitógenos y antígenos) se puede realizar en los hospitales: La Paz, 12 de Octubre, Gregorio Marañon y Universidad Complutense (Madrid), Vall d'Hebron, Sant Pau, Bellvitge (Barcelona), Virgen del Rocío (Sevilla), La Fe (Valencia), Son Dureta (Palma de Mallorca), Doctor Negrin (Canarias), Cruces (País Vasco), Reina Sofía (Córdoba), Hospital Central de Asturias y Virgen de la Arritxaca (Murcia).
5. El estudio de la función granulocitaria (NBT o test de oxidación por citometría) y de moléculas de adhesión en granulocitos se realiza en los Hospitales citados anteriormente y también en Carlos Haya (Málaga), Miguel Servet (Zaragoza) y Sant Joan de Deu (Barcelona).
6. Los estudios de la actividad micobactericida (IL-12 y IFNγ) (14) en Doctor Negrin, Vall d'Hebron, La Paz y Cruces.
7. Los estudios funcionales de todos componentes del complemento y estudios genéticos del Defecto de C1 inhibidor en Hospital La Paz (Madrid); la deleción de 28 bp en déficit de C2 de tipo I: Hospital de Cruces (Bilbao).
8. El estudio de la deleción 22q11 (síndrome De DiGeorge) 15,16 se realiza en laboratorios de genética de los hospitales: La Fe, Vall d'Hebron, Sant Joan de Deu, La Paz, Gregorio Marañón, Cruces, Virgen de la Arritxaca y Virgen del Rocío.
9. La detección de proteínas de la respuesta inmune mediante Western blot puede hacerse en La Paz y Vall d'Hebron (Btk, WASP y gp91) y Hospital General de Asturias (Btk).
10. Los estudios de mutaciones se realizan en los Centros que figuran en la tabla IV.
DISCUSION
El diagnóstico adecuado y precoz de la mayoría de IDP es esencial para iniciar las pautas terapéuticas adecuadas en cada caso, mejorar el pronóstico y orientar el estudio mutacional. Para ello, el primer paso siempre es la sospecha clínica y las primeras determinaciones para orientar el tipo de IDP. Los datos clínicos y antecedentes familiares son de gran interés. Los estudios inmunológicos básicos se realizan en la mayoría de centros hospitalarios del país. Es necesario llegar al diagnóstico del defecto genético, en las formas conocidas, para confirmar el diagnóstico inicial, realizar un pronóstico aproximado y facilitar el consejo genético familiar. Como algunas de las IDP son poco frecuentes, los diagnósticos genéticos se centralizan en unos pocos hospitales. Con la simplificación de los estudios mutacionales, habrá otros centros hospitalarios que realicen estos estudios, pero en la actualidad ya se pueden diagnosticar la mayoría de las IDP en centros españoles. Por este motivo este trabajo no pretende ser exhaustivo, sí orientativo. En las formas más graves de ID se requiere la obtención de muestras del paciente antes del tratamiento substitutivo (especialmente el trasplante de progenitores hematopoyéticos); es por lo tanto urgente realizar la sospecha diagnóstica 17 y dirigirse al centro que pueda efectuar el estudio. En algunos casos el seguimiento y la evolución clínica, así como la determinación de anticuerpos frente a gérmenes vacunales (tétanos, rubéola IgG) ayudan al diagnóstico diferencial entre hipogammaglobulinemia transitoria, procesos respiratorios por hiperreactividad bronquial y la inmunodeficiencia común variable en niños 18.
No se conocen todas las anomalías genéticas en las IDP bien catalogadas 19 y son recientes las descripciones de las IDP de la respuesta innata 20,21. El estudio de casos no identificados y sus características inmunológicas, son de gran ayuda para la definición de nuevas formas de IDP. Las consultas entre centros y profesionales dedicados a estos diagnósticos (nacionales o extranjeros) son de gran ayuda en las formas de presentación poco habituales y/o poco frecuentes.
