








Puntos clave
Un 50% de los pacientes con discinesia ciliar primaria (DCP) tienen situs inversus totalis.
Las otitis medias, la rinorrea profusa desde el nacimiento y la tos productiva crónica son los síntomas predominantes en la infancia. Las bronquiectasias y la sinusitis están presentes en la mayor parte de adolescentes y adultos. Los varones suelen ser infértiles.
El diagnóstico precoz disminuye la morbilidad y la mortalidad, y mejora el pronóstico.
La medición del óxido nítrico nasal es un método útil de cribado en pacientes con sospecha clínica de DCP. Métodos alternativos serían los estudios radioisotópicos de aclaramiento mucociliar nasal o bronquial. Todos deben confirmarse con los tests diagnósticos definitivos.
El diagnóstico definitivo exige valorar la frecuencia y el patrón de batido ciliar, así como la ultraestructura del cilio. Una ultraestructura ciliar normal no excluye la DCP.
La terapia respiratoria es extrapolada de la fibrosis quística. Puntales básicos son la fisioterapia y el control de la infección bronquial.
Los cilios son organelas celulares muy ubicuas, presentes en algún momento del desarrollo en casi todas las células. Se han conservado durante la evolución de las especies, desde los protozoos hasta los vertebrados y, aunque en el pasado se consideraban estructuras vestigiales, en los últimos años se ha descubierto su papel fundamental en numerosos procesos fisiológicos.
Habitualmente, solo pensamos en el cilio como una organela móvil, pero una gran parte de los cilios son inmóviles y actúan como antenas receptoras de muchos procesos sensoriales y como receptores de membrana, manteniendo la homeostasis de diferentes órganos y sistemas. También desempeñan un papel importante en el adecuado desarrollo embrionario.
El proceso de la ciliogénesis tiene implicaciones no solo en la morfogénesis ciliar, sino en procesos patológicos directamente relacionados con ellos.
Funciones y estructura del cilioLos cilios y los flagelos son estructuras posmitóticas que aparecen solamente en células en fases no proliferativas del ciclo celular (G0/G1 y comienzo de la fase S). Dado que la síntesis proteica no es posible en el interior del cilio, se han desarrollado proteínas de transporte intraflagelar (PTI) que, en sentido anterógrado y retrógrado, transportan las sustancias necesarias para construir el edificio ciliar. Una vez constituido, el cilio está constantemente regenerándose gracias a estas PTI. Cuando la célula va a entrar en fase G2 y M, el cilio se desmonta. Los centríolos emigran a las áreas perinucleares para formar el huso mitótico y, finalizada la división celular, vuelve a iniciarse el proceso de construcción ciliar. Es por ello que el cilio puede regular la proliferación celular a través de la modificación de distintas vías de señalización.
Los cilios son proyecciones celulares que surgen de centríolos perinucleares que emigran hacia la superficie celular o se forman allí de novo. Dan lugar al cuerpo basal, zona clave para la ciliogénesis, constituido por 9 tripletes de microtúbulos periféricos. La parte que emerge de la célula, o axonema, tiene la clásica estructura «9 + 2»: 9 pares de microtúbulos periféricos rodeando a un par central. Los flagelos, aunque de mayor tamaño, tienen una estructura similar y un papel más definido en el transporte de células y microorganismos.
El complejo tubular 9 + 2 posee una serie de conectores necesarios para la función ciliar: las uniones de nexina, que ensamblan los pares de microtúbulos entre sí y son las que mantienen al cilio intacto; los brazos radiales, que enlazan el par central, con su vaina, a los pares periféricos, y los brazos de dineína (externo e interno), que surgen de cada doblete periférico y que deslizan a los microtúbulos entre sí, y son los responsables de que el cilio se mueva (fig. 1).

Los cilios pueden clasificarse en 2 grandes tipos: móviles e inmóviles; estos últimos conocidos también como primarios o sensoriales. El cilio primario tiene una estructura 9 + 0 y es el que aparece en el nodo embrionario y en otros sistemas sensoriales, aunque se le ha considerado inmóvil y sensorial frente al 9 + 2, móvil y transportador de sustancias, no siempre se comporta así, dado que —en el embrión— estos cilios (9 + 0) son móviles y establecen la asimetría visceral (situs solitus), mientras que algunos cilios sensoriales —como los del oído interno— tienen una estructura 9 + 2 y son móviles, determinando la formación y posición de los otolitos1.
Los cilios intervienen en el desarrollo embrionario, en la polaridad de muchas células, en el mantenimiento de la homeostasis, en funciones sensoriales (oído, vista, olfato) y transportadoras, y en la división celular. Esta amplia capacidad funcional implica una gran complejidad morfológica y genética.
La función transportadora imprime movilidad a la propia célula, como sucede con el espermatozoide, o a los materiales situados sobre la superficie celular, como ocurre en las células ciliadas del aparato respiratorio, transportadoras de moco; en las de las trompas de Falopio, transportadoras del óvulo, o en las de los ventrículos cerebrales, transportadoras de líquido cefalorraquídeo. Las funciones sensoriales visuales y las homeostáticas están ligadas a las proteínas de transporte intraflagelar, que en sentido anterógrado y retrógrado transportan moléculas y proteínas2. Además, pueden actuar como mecanorreceptores, en los cilios del oído interno, por ejemplo, o como quimiorreceptores en el olfato.
La amplia distribución ciliar y su implicación funcional explican que defectos primarios del cilio desencadenen una amplísima y, en ocasiones, enigmática, variedad de manifestaciones clínicas (tabla 1) y que la disfunción, tanto de los cilios móviles como de los primarios, se liguen a una amplia gama de enfermedades conocidas como «ciliopatías»3–5:
- —
Discinesia ciliar primaria (DCP).
- —
Hidrocefalia congénita.
- —
Ceguera progresiva (retinitis pigmentaria).
- —
Hipoacusia neurosensorial (síndrome de Usher).
- —
Cardiopatías congénitas complejas, especialmente si acompañan a desórdenes de la lateralidad.
- —
Asplenia o poliesplenia.
- —
Atresia biliar.
- —
Atresia esofágica, reflujo gastroesofágico grave.
- —
Riñón poliquístico, nefronoptisis.
- —
Síndrome de Senior-Loken.
- —
Amaurosis congénita de Leber.
- —
Síndrome de Merckel-Guber.
- —
Síndrome de Joubert (síndrome cerebeloóculo- renal).
- —
Síndrome de Bardet Biedl (obesidad, hipogenitalismo, debilidad mental, defectos craneales, retinitis pigmentaria, sindactilia).
- —
Síndrome oro-facial-digital, tipo I.
- —
Síndrome de Alstrom.
- —
Síndrome de McKusick-Kaufman.
- —
Síndrome de Ellis van Creveld.
- —
JATD: condrodisplasia juvenil (distrofia torácica asfixiante).
- —
Displasia craneoectodérmica o síndrome de Sensenbrenner.
- —
Polidactilia.
- —
Heterotaxia.
Cilios en el organismo: fisiología y patología
| Cilios móviles |
| Aparato respiratorio: sistema mucociliar |
| Trompas de Falopio: arrastre del óvulo |
| Tracto cerebrospinal: circulación de líquido cefalorraquídeo |
| Flagelo del espermatozoide: transporte del mismo hacia el óvulo |
| Cilios nodales del embrión: posición visceral |
| Patología asociada |
| Discinesia ciliar primaria |
| Embarazos ectópicos |
| Hidrocefalia |
| Esterilidad masculina |
| Malposiciones viscerales, situs inversus |
| Cilios sensoriales/primarios |
| Mecano sensores en los túbulos renales |
| Receptores olfatorios |
| Receptores retinianos (conos y bastones) |
| Receptores del oído interno |
| Señalización Hedgehog (regulador de la morfogénesis y del patrón de crecimiento de diferentes órganos y tejidos) y otras vías en el desarrollo |
| Patología asociada |
| Riñón poliquístico y nefronoptisis |
| Anosmia |
| Degeneración retiniana: retinitis pigmentaria |
| Degeneración del oído interno: hipoacusia neurosensorial |
| Degeneración retiniana y oído interno: síndrome de Usher |
| Anosmia |
| Trastornos del desarrollo: obesidad, polidactilia, malformaciones del sistema nervioso central, craneofaciales, cardíacas, hepáticas, retraso mental… |
De todas ellas, la DCP es el síndrome clínico más frecuente y con mayor repercusión clínica. Se conoce también como síndrome de inmotilidad ciliar y es un trastorno hereditario, autosómico recesivo, sin predilección de sexo o raza6, que afecta a 1/10.000-60.000 individuos7,8.
La causa subyacente es una falta total de cilios
Lectura rápida
La discinesia ciliar primaria (DCP), conocida también como síndrome del cilio inmóvil, incluye diversas anomalías ciliares: aplasia, inmovilidad y discinesia ciliar, y es la forma de expresión más común de un conjunto de enfermedades conocidas como ciliopatías, a las que muchas veces está asociada.
Es una enfermedad hereditaria autosómica recesiva, en la que se han identificado 12 genes asociados. Entre ellos, las mutaciones DNAI1 y DNAH5, que codifican la dineína, se detectan en un 30–35% de los casos. Existen otros genes candidatos que codifican componentes del cilio y una variante de la DCP ligada a la retinitis pigmentaria, transmitida por el cromosoma X y causada por mutaciones del RPGR (gen regulador de la guanesina trifosfatasa de la retinitis pigmentaria). Aproximadamente el 50% de los pacientes afectados tienen situs inversus.
Lectura rápida
La alteración del aclaramiento mucociliar provoca un compromiso progresivo del tracto respiratorio superior e inferior, caracterizado por obstrucción de las vías aéreas e infecciones recurrentes del pulmón, oído medio y senos paranasales.
La forma de debut en el recién nacido es un distrés respiratorio sin causa aparente. La presencia de una rinorrea persistente desde el nacimiento, de una tos crónica, productiva, diaria o de otitis crónicas o recurrentes debe hacer sospechar esta enfermedad.
La medición del óxido nítrico exhalado nasal es un método de cribado útil en los pacientes con sospecha de DCP. Otros métodos alternativos son los estudios de aclaramiento mucociliar, nasal o bronquial, con marcadores radioisotópicos que, si son normales, excluyen la enfermedad. Ninguno de estos métodos es diagnóstico por lo que siempre, deberán ser corroborados con otros tests confirmatorios.
Tanto la discinesia como la inmovilidad conducen a una ausencia de transporte mucociliar que, en el aparato respiratorio, provoca estasis de secreciones e infección crónica de las vías respiratorias altas y bajas7,9. El trastorno de movilidad afecta también al espermatozoide y a los cilios de la trompa de Falopio, por lo que es común la esterilidad en los varones y una fertilidad reducida en las mujeres. La ineficacia de los cilios nodales embrionarios hace que la asimetría de los órganos internos se disponga al azar dando lugar, en un 50% de estos pacientes, a un situs inversus total. Su presencia, junto a sinusitis crónica y bronquiectasias, se conoce como síndrome de Kartagener, un subgrupo de la DCP con una prevalencia de 1/20.000-40.000 individuos7. En la actualidad, dado que las bronquiectasias no existen en el momento del nacimiento y que un diagnóstico precoz y la adopción de medidas terapéuticas adecuadas podrían retrasar su aparición, este síndrome podría quedar definido por la coexistencia de DCP y situs inversus10.
Manifestaciones clínicasEl diagnóstico clínico precisa de un alto índice de sospecha y el pediatra general deberá estar alerta sobre esta enfermedad, ya que al cursar con manifestaciones bastante comunes en el niño sano4,11 es fácil infravalorar su incidencia12 y retrasar su detección13. Precisamente por ello, en 2006, la Sociedad Respiratoria Europea (ERS), constituyó un grupo de trabajo (Task Force on PCD) con el fin de conocer la prevalencia y características de la DCP pediátrica, así como las actitudes diagnósticas y terapéuticas en los distintos países europeos. Sus conclusiones quedan reflejadas en 3 artículos. El primero es una normativa de consenso con las recomendaciones sobre el abordaje diagnóstico y terapéutico de la enfermedad14. El segundo recoge los factores que determinan la edad del diagnóstico en los niños europeos y corrobora que el síndrome se diagnostica escasa y tardíamente15. En el tercero, se valoran la concordancia entre la práctica clínica habitual y las recomendaciones del consenso, comprobando una gran heterogeneidad terapéutica y una pobre correspondencia con las recomendaciones de la Task Force16.
La DCP se caracteriza por una presentación precoz, desde el nacimiento, persistencia a lo largo de toda la vida y afectación multisistémica (tabla 2). Las manifestaciones clínicas son variables (tabla 3)3,14.
Manifestaciones de la discinesia ciliar primaria en los distintos órganos del cuerpo humano.
| Órgano | Manifestación clínica |
| Pulmón | Distrés respiratorio neonatal |
| Bronquitis recurrentes | |
| Bronquiectasias | |
| Oído | Otitis media secretora |
| Otitis media crónica | |
| Fosas y senos | Sinusitis crónica |
| Hipoplasia de senos, ante todo frontales | |
| Tracto genitourinario | Infertilidad masculina |
| Mujer: fertilidad disminuida, embarazo ectópico | |
| Lateralidad orgánica | Situs inversus totalis |
| Situs ambiguus (heterotaxia) | |
| Sistema nervioso central | Hidrocefalia (rara) |
Manifestaciones clínicas de la discinesia ciliar primaria (DCP) según edad.
| Órgano | Manifestación clínica |
| Prenatal | Detección por ecografía de situs inversus totalis (25% tienen DCP) o heteroataxia (prevalencia de DCP desconocida). El 40-50% de las DCP presentan situs inversus totalis |
| Ventriculomegalia cerebral fetal leve | |
| Neonatal | > 75% presentan distrés respiratorio precisando O2 |
| Rinorrea continua desde primer día de vida | |
| Situs inversus u otras formas de heteroataxia | |
| Hidrocefalia, en algunos individuos con DCP (reflejan disfunción de los cilios del epéndimo) | |
| Infancia | Tos, productiva o húmeda, crónica, asociada, o no, a atelectasias o neumonías recurrentes |
| Asma atípico, que no responde al tratamiento, especialmente si cursa con tos húmeda | |
| Bronquiectasias idiopáticas | |
| Rinitis diaria persistente. Pólipos nasales (raros a esta edad) | |
| Sinusitis crónica en niños mayores | |
| Otitis media secretora | |
| Pérdida de audición | |
| Adolescencia y edad adulta | Como en la infancia |
| Bronquiectasias más evidentes en el adulto | |
| Expectoración mucopurulenta crónica | |
| Dedos en palillo de tambor, en ocasiones | |
| Patrón espirométrico progresivo, obstructivo o mixto | |
| Poliposis nasal y halitosis | |
| Infertilidad en varones (≈50%) debida a la inmotilidad de los espermatozoides | |
| Embarazo ectópico y subfertilidad en mujeres |
Tomado de Barbato et al14.
En el período prenatal un situs inversus, detectado por ecografía, es indicio de DCP, dado que afecta al 50% de estos pacientes frente al 0,001% detectado en la población general.
En el periodo neonatal el comienzo clínico puede ser un distrés respiratorio, sin «causa aparente», con o sin neumonía12,13,17. La existencia de un situs inversus, o un antecedente familiar positivo para la enfermedad, deben orientar el diagnóstico. Una rinorrea constante, desde el primer día de vida, es un signo tan significativo de DCP17 que muchos padres indican que el niño «ha nacido resfriado».
En la infancia, es característica una tos húmeda o productiva, crónica, diaria y con expectoración mucopurulenta. Los niños pueden ser diagnosticados de asma «atípica», que no responde al tratamiento habitual3. Las otitis medias secretoras y la presencia de moco en el oído medio, en ausencia de inflamación aguda, se dan en el 85% de los casos14 y condicionan una pérdida de audición y retraso del lenguaje18. Los problemas óticos mejoran con la edad, sin desaparecer, y una hipoacusia de transmisión es la norma19,20.
En los niños mayores, es frecuente la sinusitis. La poliposis nasal es menos común que en la fibrosis quística (FQ) y en el 10% de los casos se detectan bronquiectasias14. En la adolescencia y en la edad adulta es habitual la ocupación —parcial o total— de todos los senos paranasales por tejido blando o secreciones, la hipoplasia sinusal en general y, sobre todo, la aplasia de los senos frontales (fig. 2), consecuencia de la falta del estímulo eutrófico neumatizador en la mucosa enferma. La cefalea es común, posiblemente relacionada con reagudizaciones de la sinusitis crónica, aunque también puede deberse a problemas de drenaje del líquido cefalorraquídeo21.
Lectura rápida
El diagnóstico definitivo incluye la valoración de la frecuencia y del patrón de batida ciliar, mediante vídeo de alta resolución digital y alta velocidad, así como el estudio ultraestructural del cilio. Para ello se requiere una muestra de células ciliadas procedentes de la mucosa nasal o bronquial. La toma de la muestra nasal es sencilla y rápida (apenas 2–3 s) y, raramente, requiere anestesia o sedación. Se obtiene mediante raspado, o cepillado de la mucosa, en la cara medial del tercio medio del cornete inferior. Los pacientes deben estar libres de infección respiratoria aguda desde 4–6 semanas antes, para minimizar —en lo posible— tanto las muestras con escasos cilios, como las alteraciones ciliares secundarias que complican el análisis microscópico.
Lectura rápida
Los defectos ultraestructurales se identifican en aproximadamente el 90% de los pacientes y afectan fundamentalmente a los brazos externos o internos de dineína, o a ambos. Existen defectos ciliares secundarios que pueden inducir a error y entre el 10–28% de los pacientes con DCP tienen una ultraestructura ciliar normal. Por ello, el simple análisis microscópico del cilio no es suficiente para asegurar el diagnóstico.
El diagnóstico suele ser tardío, lo que se traduce en una mayor morbilidad y peor pronóstico. El reconocimiento precoz y el inicio de un tratamiento otorrinolaringológico y pulmonar adecuado, pueden reducir y retrasar las potenciales secuelas.
Las personas afectadas de DCP pueden tener una vida normal. Aunque la infección y la inflamación bronquial son constantes y el daño pulmonar (bronquiectasias) está presente en la mayor parte de los adultos afectados, el grado de declinación de la función pulmonar es mucho más lento que en la fibrosis quística.
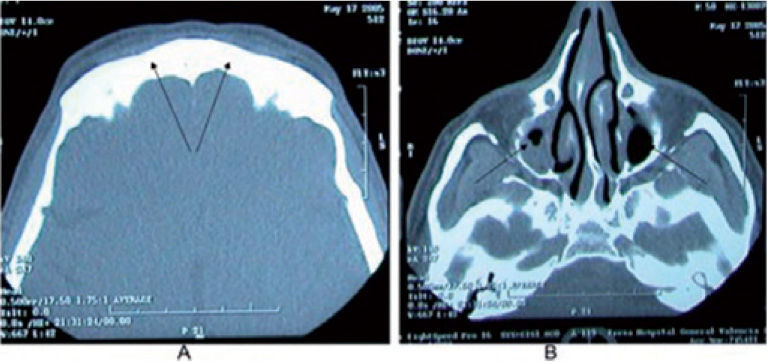
. B: hipoplasia y ocupación parcial de los senos maxilares (flecha). Estos hallazgos solo son valorables a partir de la adolescencia, dado el desarrollo cronológico de los senos paranasales.")
TCAR maxilofacual correspondiente a un paciente afectado de discinesia ciliar primaria. A: aplaxia de los senos frontales (flecha). B: hipoplasia y ocupación parcial de los senos maxilares (flecha). Estos hallazgos solo son valorables a partir de la adolescencia, dado el desarrollo cronológico de los senos paranasales.
La auscultación es frecuentemente patológica, con subcrepitantes o sibilancias que pueden emular asma. La función pulmonar permanece relativamente estable, pero hacia la tercera década de la vida suele existir un patrón obstructivo22. La tomografía computarizada de alta resolución parece ser más sensible en la detección precoz del daño pulmonar que la espirometría, sobre todo en los pacientes de mayor edad, aunque no queda clara la posible correlación entre ambas, pues mientras que en algún estudio la función pulmonar no refleja el daño pulmonar existente23, en otro, realizado en niños, la evolución de ambos parámetros es paralela24. Las anomalías radiográficas incluyen engrosamiento peribronquial, atelectasias y atrapamiento aéreo y, en la mayor parte de casos, bronquiectasias12,25. La infección bronquial se inicia precozmente y es la mayor causa de morbilidad y mortalidad en la DCP. Las bacterias más comunes en niños y adolescentes son Haemophylus influenzae no tipificable, Staphylococcus aureus y Streptococcus pneumoniae. En los pacientes mayores con enfermedad avanzada, se aísla más comúnmente Pseudomonas aeruginosa y micobacterias no tuberculosas12. La inflamación pulmonar es fundamentalmente neutrofílica, similar a la de la FQ, a pesar de que la expresión pulmonar es habitualmente más leve en la DCP26.
La mayoría de los varones tienen espermatozoides inmóviles y unos ductos eferentes testiculares sin actividad ciliar, por lo que son infértiles. Una minoría (< 20%) puede fecundar porque, aunque la estructura del cilio y del espermatozoide es similar, su composición polipeptídica es diferente27. En la mujer, la deficiente actividad de los cilios en la trompa de Falopio entorpece el transporte del óvulo hacia el útero, lo que incrementa el número de embarazos ectópicos14,28 y reduce un 50% su fertilidad.
DiagnósticoEl diagnóstico de la DCP se apoya en una clínica sospechosa, en la demostración objetiva de la disfunción o inmotilidad ciliar y en la existencia, o no, de alteraciones ultraestructurales en el cilio. Tanto la exploración funcional como el estudio ultramicroscópico del cilio exigen la utilización de técnicas específicas, no siempre al alcance de todos los centros, y la existencia de un equipo experimentado que sepa interpretarlas. En los lugares que no disponen de las mismas, se pueden plantear inicialmente unas pruebas de cribado, cuyos resultados podrían descartar ya la enfermedad, o apoyar la realización de la segunda línea de exploraciones, específicas y confirmatorias14.
Tests de cribadoMedición del óxido nítrico exhalado nasalEn la actualidad se propugna como el test de primera elección para el diagnóstico de la DCP29 (95% de sensibilidad y 90% de especificidad) 30, ya que sus valores son muy bajos o están ausentes en estos pacientes12,31, incluso en aquellos cuyos cilios tienen una ultraestructura normal32. Las cifras elevadas excluyen habitualmente la enfermedad14,31, pero no ocurre así con los valores normales30. Los valores bajos requieren realizar tests de confirmación, dado que otras enfermedades (fibrosis quística, panbronquiolitis, sinusitis crónica y poliposis nasal) también los tienen3,14. Es necesario estandarizar su determinación en todos los grupos de edad, sobre todo en los niños pequeños33. Recientemente, se han publicado valores de referencia y puntos de corte para distintas edades, utilizando 3 métodos distintos de medición: retención de la respiración (BH-NNO), exhalación oral contra la resistencia (OE-RNNO) y respiración corriente (TB-NNO), llegando a la conclusión de que el óxido nítrico exhalado nasal es una herramienta útil en todos los grupos de edad, siempre que el método aplicado se adecue a la edad del paciente29.
Medida del aclaramiento mucociliarEntre los métodos utilizados, el test de la sacarina es subjetivo, difícil de interpretar, no informativo en casos de batido discinético34, y no se recomienda en niños < 12 años14,32,35. El estudio del transporte mucociliar nasal con seroalbúmina marcada con 99TC es un test objetivo, muy específico pero poco sensible, que puede utilizarse a cualquier edad36, incluso en el recién nacido37. Su mayor mérito es que se trata de una técnica de exclusión34,38, ya que un transporte normal permite descartar la DCP, pero un test alterado no la diagnostica, dado que otras enfermedades también pueden alterarlo (discinesias ciliares secundarias). Otros estudios con radioaerosoles permiten medir el aclaramiento mucociliar pulmonar, a partir de los 5 años de edad, con una sensibilidad del 88% y una especificidad del 100%39.
Test diagnósticos definitivosNo hay ningún «gold standard» para el diagnóstico de todos los fenotipos de DCP, por lo que es necesario, tal como recomienda la Task Force on PCD14, efectuar una rigurosa evaluación tanto de la frecuencia (FBC) y patrón de batido ciliar (PBC), mediante vídeo de alta resolución digital y alta velocidad (digital high speed video [DHSV]), como de la ultraestructura del cilio, mediante microscopia electrónica de transmisión (ME). Aunque la evaluación del PBC requiere un alto grado de habilidad y experiencia, es necesario efectuarlo siempre, dado que existen casos con movimiento discinético y frecuencia de batida normal40, que no serían diagnosticados con la sola determinación de la FBC14. Además, cada vez se describen más casos atípicos de DCP con ultraestructura ciliar normal (entre un 10 y 28%)10,41,42, que serían pasados por alto en centros donde el diagnóstico depende sólo de la ME43.
Ambas técnicas requieren la toma de una muestra de células ciliadas del epitelio respiratorio, nasal (más accesible) o bronquial y, para ello, el cepillado o el raspado (más rentable), son los métodos recomendados.
Es imperativo que la muestra de células ciliadas sea correctamente manipulada para que los resultados sean fiables, ya que las células, tras su separación del epitelio, entran en apoptosis en muy poco tiempo43.
Estudio de la batida ciliar mediante «Digital high speedy video»Se efectúa analizando la muestra, depositada en un medio de cultivo celular, inmediatamente después de su obtención. Permite examinar la batida ciliar en diferentes planos, a distintas velocidades, incluso imagen a imagen44, tras su grabación con una cámara digital de alta velocidad, y la aplicación de un programa informático que mide:
- —
La FBC, considerada normal si es ≥ 9 Hz (540 batidas por minuto).
- —
El PBC, con sus 2 ciclos: fase de extensión y fase de recuperación. Es el test diagnóstico con mayor sensibilidad, especificidad, valor predictivo negativo y positivo45.
Una forma alternativa de determinar la eficacia de la batida es el test de rotación celular, que indica normalidad cuando las células son capaces de rotar sobre sí mismas en el medio de cultivo.
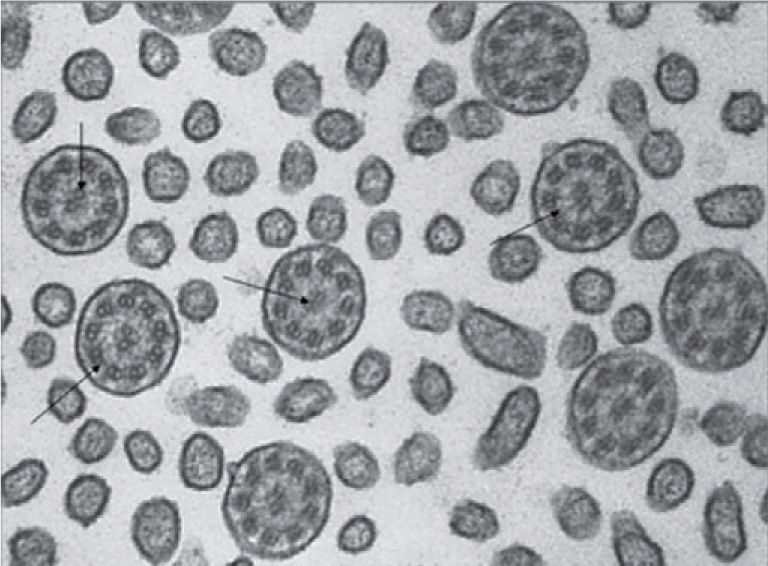
Estudio de la ultraestructura ciliar (microscopia electrónica)En este caso, la muestra se sumerge en glutaraldehído y se procesa para su estudio mediante ME. Se examinan entre 10–100 cortes transversales por paciente y se analizan tanto la orientación de los cilios, como los distintos componentes del axonema ciliar. Requiere de personal experto y criterios específicos, cuantitativos y cualitativos, sobre las distintas estructuras ciliares que se estudiarán14,42,46. Se considera que existe ausencia de dineína cuando el número medio de brazos de dineína, contados por cada corte trasversal, es menor de 2. Se definen como déficits parciales tanto a la ausencia de brazos internos (cuando el número medio es < 0,6 por corte transversal), como a la de los externos (cuando es menor de 1,6); pocos brazos de dineína (externos o internos) cuando el valor medio es menor de 7 y 3, respectivamente, y brazos cortos de dineína cuando la proyección de los mismos es corta respecto a la de los cilios normales. La orientación ciliar se considera normal si la disposición de los túbulos centrales varía menos de 28°. Las alteraciones del patrón 9 + 2 se consideran significativas cuando afectan a más del 30% de los axonemas ciliares10,42,43.
Los defectos más frecuentes (80–95% de los casos) son la ausencia completa de dineína, que se asocia a inmovilidad ciliar, y la ausencia parcial (de brazos internos o externos, poca dineína o brazos cortos), que se liga a discinesia (fig. 3). Otras anomalías incluyen: la ausencia de brazos radiales, la transposición ciliar y la agenesia de túbulos centrales3,7,42 aunque, desde un punto de vista exclusivamente morfológico, solo el déficit total de dineína se considera diagnóstico. La aplasia ciliar es un hallazgo extraordinariamente raro. En caso de duda, son útiles los cultivos celulares, pues los cilios neoformados reproducen las alteraciones congénitas, y no las adquiridas42. Una ultraestructura normal no descarta la DCP11,14.
Test genéticos
Las pruebas genéticas en la DCP son todo un reto debido a la amplia heterogeneidad y el gran número de posibles genes causantes involucrados47. Actualmente, se está intentando a nivel internacional identificar y definir los genes y las mutaciones causantes de la enfermedad. Hasta ahora se conocen 12 genes asociados a la DCP: DNAI1, DNAH5, TXNDC3, DNAH11, DNAI2, DNAAF2 [C14orf104],
Lectura rápida
La fisioterapia respiratoria, la práctica deportiva y la instauración precoz de una antibioterapia agresiva frente a las infecciones respiratorias, son la base del tratamiento. El tabaquismo activo y pasivo debe ser evitado. El papel de otros fármacos (mucolíticos, broncodilatadores, corticoides, etc.) está cuestionado.
Las intervenciones quirúrgicas para tratar la afectación del oído medio, las sinusitis o las poliposis están discutidas, pero pueden ser necesarias en algunos pacientes.
Dado que la inmovilidad o discinesia ciliar se acompañan, habitualmente, de una motilidad anormal del espermatozoide, los pacientes varones deberían ser informados de su posible infertilidad y aconsejados sobre técnicas de fertilización in vitro.
El manejo de la DCP no se apoya en evidencias científicas al carecer de estudios apropiados. La mayor parte de las recomendaciones terapéuticas extrapolan las pautas de tratamiento de la fibrosis quística3,11,14,18.
Se aconseja que el diagnóstico y el seguimiento de los pacientes con DCP sean realizados, total o complementariamente, en un centro especializado en esta enfermedad14,18,21. En cualquier caso, deben ser controlados cadencialmente por especialistas en neumología y otorrinolaringología.
La fisioterapia respiratoria y el ejercicio físico son puntales básicos del tratamiento, y la cobertura vacunal completa, incluidas las vacunas antigripal y antineumocóccica, debe plantearse en todos los pacientes. El tabaquismo, activo y pasivo, debe ser firmemente desaconsejado.
Se recomienda practicar cultivos seriados de esputo (≥ 3 meses) en orden a detectar precozmente patógenos respiratorios que exijan un tratamiento de erradicación14.
Las reagudizaciones infecciosas deben tratarse precozmente con ciclos orales, prolongados, de antibióticos, a altas dosis, según antibiograma, recurriendo a la vía intravenosa, en caso de pobre respuesta. En pacientes, con infección crónica por P. aeruginosa debe contemplarse la utilización de antiobioterapia nebulizada, a largo plazo, para reducir las exacerbaciones3,14,18.
No hay datos sobre la efectividad de agentes que reduzcan la inflamación (corticoides inhalados o macrólidos)17. No se recomienda el uso de N-acetilcisteína, y la ADNasa nebulizada (mucolítico/antineutrofílico) podría ser considerada en pacientes seleccionados. El suero hipertónico salino nebulizado puede ser beneficioso para incrementar el aclaramiento del moco14 y los broncodilatadores podrían ser utilizados en los casos en que existieran signos/síntomas de asma/hiperreactividad bronquial (HRB) y una prueba confirmada de obstrucción reversible del flujo aéreo14,18.
Las infecciones agudas del oído medio requieren antibióticos. La actitud frente a las otitis secretoras es conservadora, con valoración audiológica regular y uso alternativo de audífonos. La colocación de drenajes transtimpánicos está muy discutida, ya que no mejora la otitis, persistiendo una otorrea permanente que los obstruye, por lo que el Consenso de la ERS14 aconseja «evitarla en la medida de lo posible». Aun así, las evidencias en uno u otro sentido no están claras49.
La rinorrea crónica se trata con sonados nasales frecuentes y lavados con suero salino50. Los corticoides tópicos no parecen ser beneficiosos a no ser que exista rinosinusitis13. Las reagudizaciones sinusales son escasas y se tratan con antibióticos sistémicos. Raramente, se precisa tratamiento quirúrgico y los resultados son pobres en caso de practicarse.